
Summary of Fluid Flow Research
From: an early Bone Fluid-Flow Workshop with our updates
Remodeling in the Development of Stress Fractures
Stress fractures in bone result from repetitive loading. As such, they have often been regarded as mechanical fatigue-driven processes. However, while bone readily sustains fatigue microdamage during the course of repeated loading at the stresses or strains encountered in normal activities, it does not progress to fracture in the time course seen for the development of stress fracture. This suggests that other mechanisms drive the development of stress fractures. Histopathological data from humans and racehorses suggest that increased remodeling is a prominent early feature of stress fractures. Early increases in intracortical remodeling were observed experimentally in the rabbit tibial stress fracture model (Burr et al., 1990; Burr, 1997). This suggests a central role for increased intracortical remodeling in the pathogenesis of stress fractures. Schaffler et al. (2001) proposed that the model that best explains the development of stress fractures is that of a biologically (remodeling)-driven damage accumulation system. In this model, a stress fracture occurs as a positive feedback mechanism, wherein increased mechanical usage stimulates bone turnover, which results in focally increased bone remodeling space (porosity) and decreased bone mass. There is a wide range of factors (low level bone fatigue, altered mechanical loading, injury, cytokines, and vascular alterations) that potentially can activate local bone remodeling; all of these can occur in the development of a stress fracture. With continued loading of this focally, transiently osteopenic bone, local stresses would be markedly elevated, leading to accelerated matrix damage and failure. Fracture is the result of continued repetitive loading superimposed on the decreased bone mass caused by more, and larger, resorption spaces.
Fluid Flow in the Remodeling Response to Fatigue and Disuse
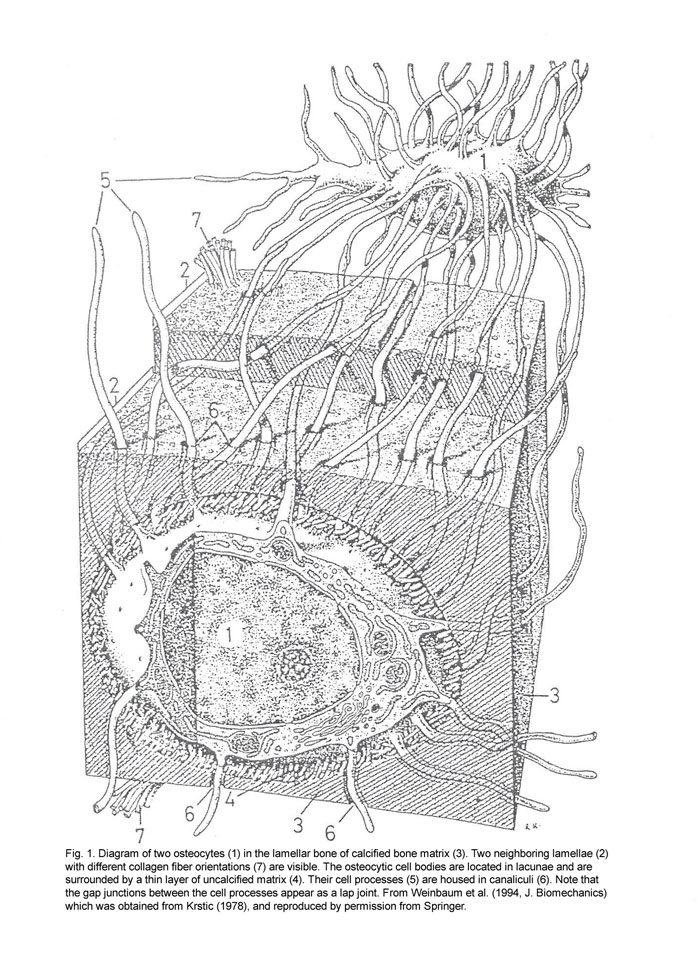
Melissa Knothe Tate and co-workers (1998) have noted that the ability of bone to regenerate itself in response to dynamic metabolic and structural demands is necessary for the survival of vertebrates. Hence, the structure of mature bone represents a patchwork; the initial construct, resulting from modeling events during growth and development, is interwoven with bone laid down by osteoblasts in areas carved out by osteoclasts during periods of remodeling. The osteocytes, which are located within the mineralized matrix of bone, are presumed to play an important role in “sensing” the mechanical and chemical environment within the tissue. Remodeling events appear to be highly “choreographed,” but the signaling and timing of interactions between osteocytes, osteoclasts and osteoblasts are not clear. Osteotropic agents are likely to mediate remodeling processes; the concentration and distribution of such agents are a function of tissue perfusion as well as diffusive and convective transport conditions prevailing in the tissue.
Knothe Tate and colleagues hypothesize that, in some cases, the reduction or loss of fluid flow and ensuing compromise to molecular transport and exchange is a mechanism causing loss of cell viability and triggering the remodeling response. Microdamage due to fatigue loading alters interstitial fluid flow and mass transport within bone, reducing the concentration and distribution of osteotropic agents to osteocytes “downstream” from the damage. In the case of disuse, the lack of fluid flow and subsequent deficiency in transport through the tissue causes the osteocytes to fall into a state of deprivation, ultimately also likely resulting in a loss of viability. Based on these concepts, a model for mechanochemical transduction in bone has been developed by these investigators.
Bone Perfusion/Reperfusion and Bone Remodeling
Stress fractures have been proposed to arise from repetitive activity of training, inducing an accumulation of microdamage (e.g., microcracks) in locations of peak strain. However, stress fractures most often occur long before accumulation of material damage could occur; they can occur in cortical locations of low, not high, strain, and intracortical osteopenia precedes typical microcracks (see last paragraph in this section regarding use of confocal microscopy to detect microdamage entities that are smaller than microcracks). Alternatively, Otter and colleagues (1999) have proposed that this lesion arises from a focal remodeling response to site-specific changes in bone perfusion during redundant axial loading of appendicular bones. Intramedullary pressures significantly exceeding peak arterial pressure are generated by strenuous exercise, and if the exercise is maintained, the bone tissue can suffer from ischemia caused by reduced blood flow into the medullary canal and therefore to the inner two-thirds of the cortex. Site specificity is caused by the lack (in certain regions of the cortex) of fluid flow, which brings nutrients from the periosteal surface to portions of the cortex. Upon cessation of the exercise, re-flow of fresh blood into the vasculature leads to reperfusion injury, causing an extended no-flow or reduced flow to that portion of the bone most strongly denied perfusion during the exercise. This leads to a cell-stress-initiated remodeling which ultimately weakens the bone, predisposing it to fracture.
It must be emphasized that, in recent years, investigators have recognized various forms of microdamage that cannot be detected with conventional light microscopy. For example, using confocal microscopy, the relatively smaller, less obvious, microdamage entities have been identified. These include osteocyte dendritic disruption, diffuse matrix damage, and wisp microcracks (Boyce et al., 1998; Herman et al., 2010; Skedros et al., 2011).
Cellular Aspects of Mechanotransduction and Bone Remodeling
Elisabeth Burger and colleagues (1999; 2000) have investigated cellular aspects of mechanotransduction and bone remodeling. The capacity of bone tissue to alter its mass and structure in response to mechanical demands has long been recognized, but the cellular mechanisms remain poorly understood. Burger and colleagues have noted several lines of evidence supporting the role of osteocytes as the principal mechanosensors of bone, and that this is mediated at the level of lacunar-canalicular porosity. Strain-derived flow of interstitial fluid through this porosity is thought to mechanically activate osteocytes. The narrowness of the canalicular annulus ensures that even the minute physiological bulk strains in bone produce considerable fluid shear stress over the osteocyte cell “dendrites.” Extracellular and/or intracellular signaling between activated osteocytes and the osteoclasts/osteoblasts at the bone surface provides a mechanism for strain-regulated modulation of bone mass.
However, to fully explain mechanical adaptation, a cellular mechanism must exist whereby not only the density of bone tissue is regulated, but also the alignment of the trabeculae and osteons along the dominant loading directions. This bespeaks of a mechanism that guides the direction in which osteoclastic resorption proceeds during the process of bone remodeling. Finite element analysis of remodeling bone at the microscopic, supra-cellular scale showed opposite strain levels around the cutting- and closing cone of a (hemi-)osteon loaded in the longitudinal (i.e. dominant) direction. A region of decreased strain appeared in front of the cutting cone of the osteon tunnel, where osteoclasts are activated to continue resorption. Likewise in a trabecula, the presence of a Howship’s lacuna induced decreased strain, along the trabecular surface in the direction of loading. Around the closing cone of an osteon tunnel however, where osteoblasts are recruited to refill the gap, elevated strains occur in the tunnel wall. Elevated strains also appeared at the bottom of the Howship’s lacuna. These associations have led these investigators to suggest that in remodeling bone, osteocytes, by being locally reduced strain fields, may guide the osteoclasts to resorb bone in the ‘right’ direction, thereby ensuring correct alignment of the new (hemi-)osteon. They also may determine, based on locally elevated strain fields, the amount of subsequent bone formation by osteoblasts, and thereby the final density of the remodeled piece of bone. Local regulation of bone metabolism by mechanically informed osteocytes therefore provides a mechanism that explains both aspects of mechanical adaptation, appropriate bone density and appropriate bone alignment.
Fluid Flow During Osteoid Tunneling
Smit and Burger (2000) have found evidence that BMU-coupling during bone remodeling may be regulated by deformation of the bone matrix under mechanical loading. Further, it is generally assumed that mechanosensing by osteocytes is related to extracellular canalicular fluid flow, which is generated by deformation of the bone matrix under mechanical loading. In an attempt to relate the theory of canalicular fluid flow to BMU coupling, they determined the pattern of fluid flow around a tunneling osteon under axial loading. They approached the problem with Biot’s theory of poroelasticity and the finite element (FE) method. The tunneling osteon was modeled axisymmetrically as a cylindrical gap with a spherical end, and the bone matrix was described as an isotropic material with a fully saturated lacunar-canalicular porosity of 5.0%. They derived the material properties of the mineralized bone matrix from the isotropic description of cortical bone by Cowin & Sadegh (1976). The bulk modulus of the bone fluid was that of water. An important parameter in the model is the hydraulic permeability k, which varies over several orders of magnitude in the literature. Their value of 1.2e-7 was two orders of magnitude smaller than the one derived by Zhang et al. (1998). The value of this parameter was discussed in some detail, and a parameter study was presented. The FE analysis showed that during a walking cycle (4 km/h) a different fluid flow pattern exists near the cutting cone as compared to the closing cone. Or in other words: the findings of this study suggest that the osteocytes within the bone matrix sense different patterns of mechanical stimulation near sites of osteoclastic and osteoblastic activity. This is compatible with the hypothesis that local patterns of bone fluid flow regulate BMU-coupling.
Bone Cells as Mechanosensory Cells
It has been proposed that osteocytes are the single mechanosensory cell type because they are ideally situated to sense mechanical stimulation, such as strain or interstitial fluid flow, as a result of mechanical loading. While there is little argument that osteocytes are subject to fluid flow, they are by no means the only bone cell type to experience hydrodynamic forces. Osteoblasts and bone lining cells are subject to intercellular fluid flow, as are osteoclasts and their precursors. Consistent with this view, all bone cell types investigated respond to fluid shear stress. The significant distance between most osteocytes from the appositional and resorption surfaces of bone further suggest that the ‘more distant’ osteocytes do not have the dominant role in load-induced remodeling.
The Role of Gap Junctions
Physical signals, in particular mechanical loading, are clearly important regulators of bone turnover. Indeed, the structural success of the skeleton is due in large part to the bone’s capacity to recognize some aspect of its functional environment as a stimulus for achievement and retention of a structurally adequate morphology. However, while the skeleton’s ability to respond to its mechanical environment is widely accepted, identification of a reasonable mechanism through which a mechanical “load” could be transformed to a signal relevant to the bone cell population has been elusive. In addition, the downstream response of bone cells to load-induced signals is unclear. Evidence suggesting that gap junctional intercellular communication (GJIC) contributes to mechanotransduction in bone and, in so doing, contributes to the regulation of bone cell differentiation by biophysical signals was reviewed. In that context, mechanotransduction is defined as transduction of a load-induced biophysical signal, such as fluid flow, substrate deformation, or electrokinetic effects, to a cell and ultimately throughout a cellular network. Thus, mechanotransduction would include interactions of extracellular signals with cellular membranes, generation of intracellular second messengers, and the propagation of these messengers, or signals they induce, through a cellular network. Henry Donahue and colleagues (Loiselle et al., 2013; Lloyd et al., 2014) have proposed that gap junctions contribute largely to the propagation of intracellular signals.
The Relationship Between Bone Fluid Flow and Adaptation
The motion of intracortical fluid flow, which arises from mechanical loading, has been proposed to be an important mediator for regulating bone mass and morphology (Li et al., 2014). However, mechanical deformation-generated stimulation may only partially explain the mechanism of flow-induced adaptation, because loading of bone results in not only intracortical fluid flow by the sources of matrix deformation and intramedullary (IM) pressure, but also the matrix strain which has been proposed as a key for the remodeling process. It has been demonstrated that bone fluid flow and its associated streaming potentials can be significantly influenced by the dynamic IM pressure that can be controlled quantitatively. The hypothesis of fluid-induced bone adaptation has been evaluated by YX Qin, Clinton Rubin and colleagues (2003) in an avian ulna model using IM hydraulic loading in the absence of bone matrix strain. The fluid pathways in bone during the loading were discussed. The left ulnae of adult male turkeys were functionally isolated via transverse epiphyseal osteotomies. A specially designed fluid-loading device was firmly attached on bone via a 5-mm hole allowing IM pressure oscillation in the cavity. A sinusoidal fluid pressure was applied to the ulna with the magnitude of 50 mm Hg, 20 Hz, 10 min/day for 4 weeks. With IM pressure generating a spatial fluid pressure gradient distribution through the cortex, fluid loading (n=4) resulted in significant new surface bone formation (12.2 ± 4.2%). In the animal group subject to sham disuse alone (n=4), the cortex showed a decrease in cross-sectional area with 6.1 ± 3.0% reduction compared to the contralateral control. The results show that low magnitude IM pressure can initiate a spatial fluid flow in bone and thus stimulate a bone adaptive response. This suggests that oscillation of IM pressures may influence the perfusion of bone tissue in many ways, e.g., altering blood supply and enhancing pressure gradients in a variety of fluid channels. IM pressure loading can increase and improve this perfusion process. Moreover, it assumes that there is a fluid pathway directly connected between the marrow cavity and intracortical porous space, e.g., Haversian canal and lacunae-canaliculi, which may play a role in regulating fluid transportation and perfusion in bone. These experiments yield insight into the mechanisms, at least at the tissue level, by which bone fluid flow initiates and controls bone morphology.
Load-Induced Fluid Flow and Functional Adaptation
R. Steck, Knothe Tate and colleagues (2003) hypothesize that load-induced fluid flow through bone enhances transport of substances (i.e. nutrients and osteotropic substances such as signal molecules, molecular factors and hormones) that modulate cellular activities associated with growth, adaptation and repair, thereby providing a mechanochemical transduction mechanism for functional adaptation. In order to start to understand the implications of fluid flow for processes associated with remodeling, they have developed a macroscopic continuum model of the rat tibia in parallel with molecular tracer experiments using an established four-point-bending model of the rat tibia. They used a two-step finite element (FE) approach to calculate different fluid flow parameters on a macroscopic scale. In a first step, bone was modeled as a poroelastic continuum for the calculation of fluid velocities and displacements resulting from the loading schemes of the experimental models. In a second step, these fluid velocities were used in a mass transfer analysis to demonstrate the positive effect of this additional convective flux on the distribution of a simulated tracer within bone cross-sections. The purpose of this presentation was to introduce the theoretical model and to interpret the predictions of the model in light of experimental data from the molecular tracer experiments as well as from parallel experiments in which a functional adaptation response was elicited in response to the hyperphysiological loading regime.
Fluid Flow Induced Strain Amplification on Bone Cells
Cell processes of osteocytes in their canaliculi provide a greatly simplified and heretofore unexploited model system to explore the effect of fluid drag forces on extracellular matrix, its coupling to the intracellular actin cytoskeleton (IAC), and the strain amplification that results from this coupling. The model also provides a resolution to a fundamental paradox in bone physiology; namely, that the strains applied to whole bone (i.e., tissue level strains) are much smaller (0.04% to 0.3%) than the strains (1% to 10%) that are necessary to cause bone signaling in deformed cell cultures. The model of You and colleagues shows that:
- 1. it is indeed possible to produce cellular level strains in bone that are more than 100-fold greater than tissue level strains, and
- 2. the fluid-flow-induced drag forces on the fibers that tether the cell to its surrounding extracellular matrix can be much greater than the fluid shear forces on the cell membrane; the fluid force that has been extensively studied until now.
Additional information can be found in the works of S. Weinbaum, L. You, and colleagues (2001a; 2011).
The Importance of Flow Reversal in the Response of Bone Cells to Fluid Flow
Loading-induced fluid flow in the lacunar-canalicular system is oscillatory in nature due to the dynamic nature of most physical activities. Thus, it is important that the in vitro investigations of loading-induced fluid flow as a physical signal regulating bone cell metabolism also is oscillatory and involve a reversal of flow direction. For example, Chris Jacobs and coworkers (2001b; 2010) have found that GdCl (a putative stretch-activated channel blocker) had no effect on cytosolic calcium mobilization and gene expression in response to oscillatory flow, but does have an effect in the response to steady flow.
Osteopontin and Bone Remodeling
The non-collagenous protein, osteopontin (OPN), has the properties of a cytokine but will also promote cell attachment to mineralized matrices. The protein is found in bone and body fluids; it interacts with receptors (integrins, possibly CD44v) to stimulate intracellular signaling pathways that control gene expression and cell behavior. It is chemotactic for macrophages and in some situations aids cell survival, likely by inhibiting apoptosis. Research on OPN-deficient (knock-out) mice has revealed that OPN is required for cell-mediated immunity and for bone remodeling in response to stress. Masaki Noda (2006) described some of the studies in his laboratory on the OPN-deficient mice described by Rittling et al. (1998). In contrast to control animals, ovariectomized OPN-deficient mice retain most of their bone mineral. Additionally, the knockout mice are resistant to disuse osteoporosis, and are much less efficient at resorbing ectopic bone. It appears that in the absence of OPN osteoclast function is impaired.
References
Boyce TM, Fyhrie DP, Glotkowski MC, Radin EL, Schaffler MB. 1998. Damage type and strain mode associations in human compact bone bending fatigue. J. Orthop. Res. 16:322-329.
Burger EH, Klein-Nulend J. 1999. Mechanotransduction in bone — role of the lacunocanalicular network. FASEB J. 13:S101-S112.
Burr DB. 1997. Bone, exercise, and stress-fractures. Exerc Sport Sci. Rev. 25:171-194.
Burr DB, Milgrom C, Boyd RD, Higgins WL, Robin G, Radin EL. 1990. Experimental stress fractures of the tibia. Biological and mechanical aetiology in rabbits. J Bone Joint Surg Br 72:370-375.
Cowin SC, Hegedus DH. 1976. Bone remodeling I: Theory of adaptive elasticity. J Elasticity 6:313-326.
Herman BC, Cardoso L, Majeska RJ, Jepsen KJ, Schaffler MB. 2010. Activation of bone remodeling after fatigue: differential response to linear microcracks and diffuse damage. Bone 47:766-772.
Ishijima M, Ezura Y, Tsuji K, Rittling SR, Kurosawa H, Denhardt DT, Emi M, Nifuji A, Noda M. 2006. Osteopontin is associated with nuclear factor kappaB gene expression during tail-suspension-induced bone loss. Exp Cell Res 312:3075-3083.
Jacobs CR, Temiyasathit S, Castillo AB. 2010. Osteocyte mechanobiology and pericellular mechanics. Annu Rev Biomed Eng 12:369-400.
Knothe Tate ML, Knothe U, Niederer P. 1998. Experimental elucidation of mechanical load-induced fluid flow and it’s potential role in bone metabolism and function adaptation. Am. J. Med. Sci. 34:189-195.
Li P, Liu C, Hu M, Long M, Zhang D, Huo B. 2014. Fluid flow-induced calcium response in osteoclasts: signaling pathways. Ann Biomed Eng 42:1250-1260.
Lloyd SA, Loiselle AE, Zhang Y, Donahue HJ. 2014. Shifting paradigms on the role of connexin43 in the skeletal response to mechanical load. J Bone Miner Res 29:275-286.
Loiselle AE, Jiang JX, Donahue HJ. 2013. Gap junction and hemichannel functions in osteocytes. Bone 54:205-212.
Otter MW, Qin YX, Rubin CT, McLeod KJ. 1999. Does bone perfusion/reperfusion initiate bone remodeling and the stress fracture syndrome? Medical Hypothesis 53:363-368.
Qin YX, Kaplan T, Saldanha A, Rubin C. 2003. Fluid pressure gradients, arising from oscillations in intramedullary pressure, is correlated with the formation of bone and inhibition of intracortical porosity. J Biomech 36:1427-1437.
Rittling SR, Matsumoto HN, McKee MD, Nanci A, An XR, Novick KE, Kowalski AJ, Noda M, Denhardt DT. 1998. Mice lacking osteopontin show normal development and bone structure but display altered osteoclast formation in vitro. J Bone Miner Res 13:1101-1111.
Schaffler MB. 2001. Bone Fatigue and Remodeling in the development of stress fractures. In: Burr DB, Milgrom C, editors. Musculoskeletal fatigue and stress fractures. Boca Raton: CRC Press. p 161-182.
Skedros JG, Sybrowsky CL, Anderson WE, Chow F. 2011. Relationships between in vivo microdamage and the remarkable regional material and strain heterogeneity of cortical bone of adult deer, elk, sheep and horse calcanei. J Anat 219:722-733.
Smit TH, Burger EH. 2000. Is BMU-coupling a strain-regulated phenomenon? A finite element analysis. J Bone Miner Res 15:301-307.
Steck R, Niederer P, Knothe Tate ML. 2003. A finite element analysis for the prediction of load-induced fluid flow and mechanochemical transduction in bone. J Theor Biol 220:249-259.
Weinbaum S, Duan Y, Thi MM, You L. 2011. An Integrative Review of Mechanotransduction in Endothelial, Epithelial (Renal) and Dendritic Cells (Osteocytes). Cell Mol Bioeng 4:510-537.
You J, Cowin SC, Schaffler M, Weinbaum S. 2001a. A model for strain amplification in the actin cytoskeleton of osteocytes due to fluid drag on pericellular matrix. J. Biomech 34:1375-1386.
You J, Reilly GC, Zhen X, Yellowley CE, Chen Q, Donahue HJ, Jacobs CR. 2001b. Osteopontin gene regulation by oscillatory fluid flow via intracellular calcium mobilization and activation of mitogen-activated protein kinase in MC3T3-E1 osteoblasts. J Biol Chem 276:13365-13371.
Zhang D, Weinbaum S, Cowin SC. 1998. Estimates of the peak pressures in bone pore water. J Biomech Eng 120:697-703.