Bone Dysplasias
A. Intro
a. Dysplasia is defined as a generalized developmental abnormality occurring because of a pathologic organization of cells within a specific tissue type. There are greater than 200 described skeletal dysplasias.
b. Many classifications have been put forth, classically they were based on pattern of involvement. Newer classifications are more often based on etiology, i.e. genetic defect, abnormal protein or enzyme.
c. Terminology
i. Dwarfing condition – disproportionately short stature.
1. short-trunk
2. short-limb
a. rhizomelic – shortening of the root (proximal) portion
b. mesomelic – shortening of the middle portion
c. acromelic – shortening of distal portion
ii. diastrophic – to grow twisted
iii. camptomelic – bent limbs
This chapter is divided into two main sections, the first being a lot of material borrowed from Dr. Michael Richardson from UW who nicely outlines a common sense approach to common dysplasias. His information is supplemented with pictures and discussion of common treatments for the given dysplasias. The second section then mops up other ‘fairly common’ dysplasias that he didn’t mention. Much of this information was “borrowed” from multiple other sources including:
Morrissy R & Weinstein S: Lovell and Winter’s Pediatric Orthopaedics, 5th
edition. Philadelphia, LL&W, 1993
Stefko R & Erickson M: Pediatric Orthopaedics in Miller’ Review of Orthopedics,
3rd Edition. Philadelphia, Saunders, 2000.
Chapman M: Chapman’s Orthopaedic Surgery, 3rd edition. Philadelphia, LL&W,
1993
http://www.rad.washington.edu/mskbook/dysplasia.html
A Quick & Dirty Approach to Skeletal Dysplasias
My personal approach has these five simple steps:
1. The first thing is to somehow tumble to the idea that the patient might have a dysplasia.
2. Next, check and see if the findings are due to some acquired problem that you already know a lot about — if so, life is good.
3. OK, sigh… it’s not an acquired disorder. Run through your mental short list of commonly seen congenital dysplasias — if this patient is on it, life can still be good.
4. If the patient isn’t on the short list, look for the child in Taybi and Lachman’s seminal book.
5. If you are still uncertain, send the patient to an expert for another opinion. Even if you are pretty sure of the diagnosis, this still may not be a bad rule.
1. Could it be a dysplasia?
This first step may be the hardest one. There are a lot of physicians out there in deep denial about dysplasias. They dislike so much the idea of having to do a dysplasia workup that they may refuse to even put dysplasia on their mental lists. As soon as they consider the “D” word, up come visions of bird-headed dwarves and hand-foot-uterus syndrome. I call this the mental Moro reflex. My advice: get over it — bird-headed dwarves and hand-foot-uterus syndrome are quite rare (I’m still waiting to see my first case of either). Most cases of dysplasia that I see are usually due to some common acquired disorder or some relatively common congenital dysplasia.
To be a bit more charitable to my colleagues, sometimes we also tend to focus so tightly on one particular finding and how to explain it that we miss the big picture. One handy clue to recognizing dysplasias is to note that the process is systemic or generalized, rather than focal. Another handy clue is to recall what the word dysplasia really means. Dysplasia comes from Latin roots dys and plasia, meaning “bad growth”. Therefore, if your patient has bones that are funny-shaped in some way, they are probably dysplastic. As you look at a case, if you find yourself thinking words like “strange”, “bizarre”, “peculiar”, “weird”, etc. train your brain to automatically translate this into dysplasia, dysplasia, dysplasia,….
2. Could this be an acquired process?
Don’t forget that there are also acquired causes of dysplastic bones, of course. This simple fact saves my personal butt about half the time when I’m asked to consult on someone with bizarre-looking bones. Acquired disorders are much more common than congenital dysplasia in the patient population I usually see, even in the big tertiary medical center in which I practice. Not only that, but unusual manifestations of common, acquired disorders are still more common than congenital dysplasias in my practice. Unless you end up practicing in some large center that specialized in dysplasias, your experience will probably be a lot like mine.
In order for growing bones to end up with a normal shape and size, they need normal muscle pull and gravitational loading while they are growing. Therefore, any process that interferes with this normal loading may lead to funny looking bones. Paraplegia or quadriplegia are extreme examples of this. Childhood paraplegics may end up with extremely gracile (long and thin) bones, with coxa valga, a small and funny looking pelvis, and very little muscle mass. On the other end of the spectrum, any chronic disease that keeps a child bedridden a lot during their growth phase (leukemia, juvenile chronic arthritis, hemophilia, etc.) may give them mildly dysplastic bones that look like one of the neuromuscular diseases, such as paraplegia. Fractures that heal poorly or which have had extensive surgery can appear quite bizarre at times, and should be on the differential for acquired bone dysplasias. So, this is where history comes in handy. Once you have gotten the clinical lowdown, you should be able to refine the diagnosis a bit more.
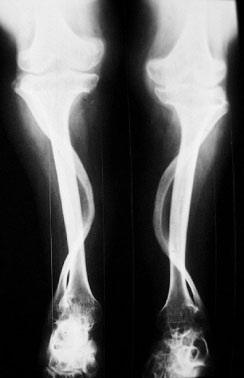
A patient with rather dysplastic fibulas due to juvenile rheumatoid arthritis. The proximal tibias are also a bit odd due to a prior bilateral high tibial osteotomies.
3. Could this be a common dysplasia?
Specialty books list hundreds of different types and subtypes of skeletal dysplasias. This is surely heady wine for the connoisseur of such things. However, the rest of us would love to have a short list of the ones we’re most likely to see. Some guidance can be gained from a recent compilation by Kozlowski and Beighton. These authors listed every dysplasia that they could think of and tried to rank each from 0 to 4 stars, corresponding to how common it was. A four star ranking meant that there are over 1000 cases of it reported in the literature. Three stars means that there are between 100 and 1000 cases, and so on. I agree with most of their rankings, but would adjust some of the entities a bit. My modified Short List Of Dysplasias (SLOD) is listed here in alphabetical order:
* Achondroplasia
* Cleidocranial dysostosis
* Dactyly
* Brachydactyly
* Camptodactyly
* Polydactyly
* Syndactyly
* Enchondromatosis (Ollier’s)
* Fibrous dysplasia
* usual form (Jaffe-Lichtenstein)
* with skin pigmentation and precocious puberty (McCune-Albright)
* Gaucher’s
* Hypophosphatemic rickets
* Marfan’s
* Multiple hereditary exostoses
* Neurofibromatosis
* Osteogenesis imperfecta
* Osteopetrosis
* (with osteopetrosis, you get pyknodysostosis for free)
* Osteopoikilosis
I dropped this list into some anagram maker software, and it burped out over 300 possible mnemonics. Among the more intriguing were: COMMON HEAD FOG (very common after studying dysplasias), COMMON DEAF HOG (we all feel like this at times), HO! COG OF MADMEN (senior residents approaching boards time), and MACHO MEN OF GOD (most people never get this confident about dysplasias). So, I leave it to the reader to choose the mnemonic that sticks in your brain the best.
Having regurgitated this list of possibilities, it is time to check and see if the gods are smiling, and our patient has one of these entities. Unfortunately, to do this, you actually have to learn something about these entities, so gird your loins and do so. Listed below are a few germane facts about each of them to get you started.
Achondroplasia
First, a few words on dwarfism. Most types are very rare and quite a few are lethal. Of the non-lethal types, the only really common type of short-limbed dwarfism is achondroplasia. Therefore, once you have learned the important stuff about achondroplasia, the smart money suggests moving on to more practical topics.
Inheritance: AD, 80-90% from new mutation, caused by point mutation in the gene for
fibroblast growth factor receptor-3 (FGFR3), on chromosome 4
Frequency: 1/15-40,000
Clinical: Prototype of short-limbed dwarfism (rhizomelic), 118-145 cm (male)/112-136 cm (female) adult height; frequent middle ear infections, dental crowding; C-section required to prevent subarachnoid hemorrhage as hydrocephalus incidence is 3% (head size normally >97%); prominent frontal bossing, trident hand, obesity common. Significant respiratory problems in 10% because of abnormal thoracic cage configuration; midface hypoplasia, upper airway obstruction, or spinal cord compression at foramen magnum (central sleep apnea, respiratory dysfunction, cyanotic episodes, feeding problems, quadriparesis, sudden death, delayed ambulation).
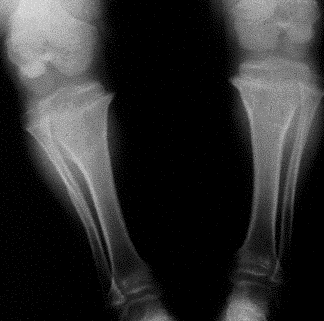
Radiographic: Short pedicles and interpedicular narrowing from L1-L5; frequent TL kyphosis; metaphyseal flaring; small foramen magnum; disproportionately long fibula; iliac wings underdeveloped with flat acetabular roof.
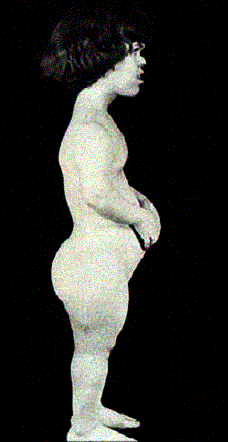
Pathology: Physis is shorter than normal due to deficit in proliferative stage of endochondral ossification—due to FGFR3 ‘gain-of-function’ mutation. Receptor normally involved in regulation of growth by suppressing proliferation. FGFR3 mutation causes over-suppression of growth in endochondral ossification.
Prognosis: Homozygous achondroplasia is generally lethal, life expectancy is not significantly diminished in heterozygotes.
Treatment: Limb lengthening is controversial; overgrowth of fibula may or may not be associated with genu varum requiring tibial osteotomy or fibular epiphysiodesis—not responsive to bracing; cervical and lumbar laminectomy for spinal stenosis is common; surgical enlargement of foramen magnum is indicated for clonus, hyperreflexia or central sleep apnea; TL kyphosis resolves in 90% with walking but if persists treatment with extension orthoses and beyond age 5 surgery for curve > 40-60 degrees.
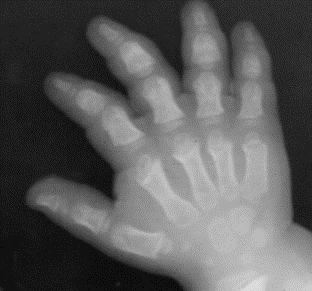
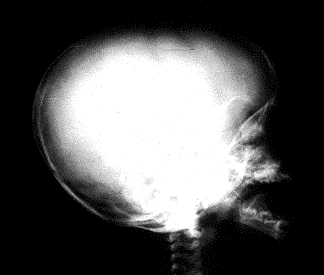
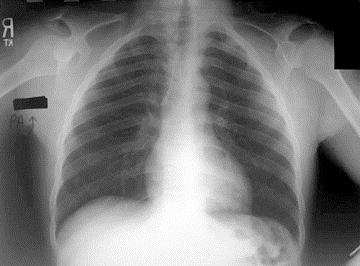
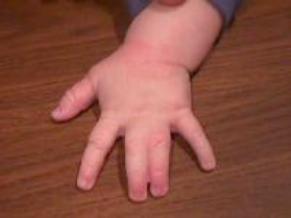
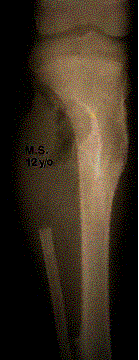
Images above demonstrates shortened phalanges (especially proximal) and classic “trident hand” with space between 3rd and 4th fingers. Right-hand image illustrates normal-sized cranium with small facial structure.
Cleidocranial Dysostosis (aka cleidocranial dysplasia)
This is an autosomal dominant disorder whose very name tells us a lot about it. Dysostosis indicates an abnormality in the development of bone, and cleido- (clavicle) and cranial (head) tell us where major abnormalities occur. This disorder occurs in both membranous and endochondral bone, and has a striking propensity for affecting midline structures. If you painted a big, broad stripe down the midline with a paintbrush from skull to groin, you’d paint over a lot of structures involved with this syndrome.
Inheritance: AD, several chromosome abnormalities have been reported to be associated with this syndrome, including rearrangement of long arm of chromosome 8 (8q22) and the long arm of chromosome 6 (band p21), coding for RF CBFA1 (core-binding factor alpha subunit 1), which encodes a TF required for osteoblast differentiation.
Frequency:1:200,000
Clinical: A congenital disorder of bone formation with clavicular hypoplasia or agenesis with a narrow thorax, allowing approximation the shoulders in front of the chest occurring with delayed ossification of the skull, excessively large fontanelles, and delayed closing of the sutures. The fontanelles may remain open until adulthood, but the sutures often close with interposition of Wormian bones. Bosses of the frontal, parietal, and occipital regions give the skull a large globular shape with small face. The characteristic skull abnormalities are sometimes referred to as the “Arnold head” named after the descendants of a Chinese who settled in South Africa and changed his name to Arnold. More than 100 additional anomalies may be associated, including wide pubic symphysis, dental abnormalities, short middle phalanges of the fifth fingers, delayed skeletal maturation, hearing deficiency, and mild mental retardation in some cases.
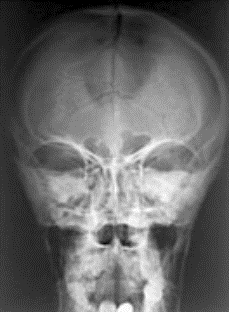
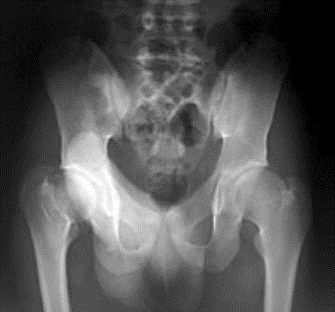
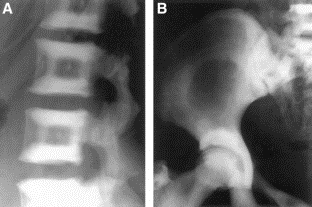
Note the delayed closure of the sagittal suture, as well as several Wormian bones. The pelvis exhibits narrow iliac wings, hypoplasia of the pubic bones, and bilateral coxa vara.
Radiographic: Pelvis: Delayed closure of wide symphysis pubis. Hand and foot: Pseudoepiphyses at the base of the metacarpal bones, abnormal phalangeal tufts of the hands and feet, short middle phalanges of the fifth fingers, and cone-shaped epiphyses of the distal phalanges. Extremities: Coxa vara, coxa valga, and notching capital femoral epiphysis. Spine: Spina bifida, syringomyelia, and spondylolysis.
Prognosis: These patients have a normal life expectancy. Prominent complications of this syndrome include dental anomalies, hearing loss, scoliosis, and dislocations of the shoulder, radial head or hip.
Treatment: No treatment indicated for clavicles. Consider intertrochanteric valgus osteotomy if neck-shaft angle is
Dactyly
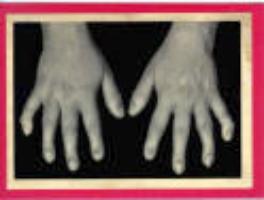
Various abnormalities of the fingers may be seen, either alone or in association with other findings in a variety of syndromes. If you spot funny looking fingers, characterize them and then look in Drs. Taybi and Lachman’s book for zillions of possible causes. How do you characterize them? English will do — short fingers, fused fingers, and too many fingers are common varieties. However, if you want to look these up in a gamuts book, you first have to translate this to Doctor Talk. Ergo, a short glossary follows:
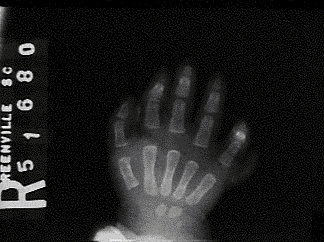
English Doctor Talk
Short fingers Brachydactyly
Too many fingers Polydactyly
Two or more fingers are fused Syndactyly
Contractures of fingers Camptodactyly
Inclined fingers, usually fifth Clinodactyly
Long, spider-like fingers Arachnodactyly
Associated Syndromes:
Poland’s syndrome:
– hypoplasia of hand and simple syndactyly of fingers on the same
side as the absent pectoral muscles (and other chest wall muscles);
Apert’s Syndrome:
– when all digits are joined, as is common in spoon hand of Apert’s
syndrome (acrocephalosyndactyly), it is important to release
border digits-thumb and small finger-first;
– remaining 3 joined fingers can be managed by removing middle digit,
thus creating a three-fingered hand with a thumb and sufficient
skin for closure
Treatment:
Syndactyly: (most common hand anomaly) Usually released at about 1 yr of age (acrosyndactyly in neonatal period). Shouldn’t separate both sides of same digit in same sitting due to circulatory problems. The new web is constructed with local skin, full-thickness grafts are almost always required.
Polydactyly: May require soft tissue or more complex bony reconstruction.
Enchondromatosis
Most enchondromas are solitary. However, some unfortunate patients may have a syndrome of multiple enchondromas, a.k.a. Ollier’s syndrome. This syndrome, unlike MHE (see below) is not hereditary.
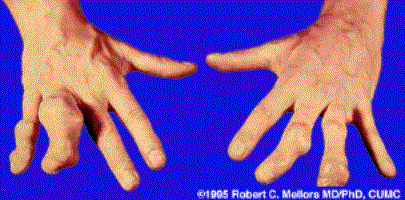
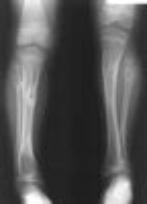
Clinical: The condition usually presents between 2 and 10 years of age. The most common site for enchondromas is the hands, followed by the feet and forearms. The skull, spine, and breastbone are seldom affected
Radiographic: Along with the classic central expansile pattern seen with classic solitary enchondromas, one may also see linear or columnar lucencies in the metaphyses, representing columns of growing cartilage.
Prognosis: The main significance of this disorder is that some lesions will undergo malignant degeneration (5 – 30 %), usually to chondrosarcoma. This likelihood is even higher (approaches 100 %) when multiple enchondromas are associated with soft tissue hemangiomas (Maffucci’s syndrome). These hemangiomas may contain phleboliths, making the diagnosis possible on plain radiographs. The pelvis and shoulder girdle are the most common locations of secondary chondrosarcoma.

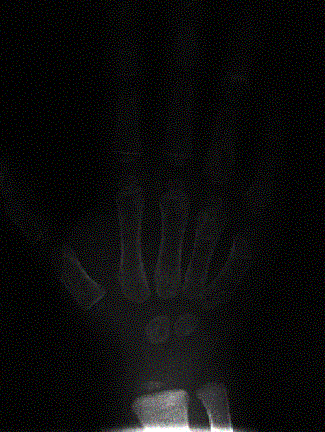
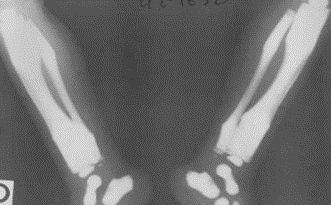
Multiple lucent metaphyseal lesions of the proximal and middle phalanges of the fourth and fifth digits, as well as the distal metacarpals of the fourth and fifth digits.
Treatment: Extremity deformities should be managed surgically to maintain the function of the limbs without specific regard to the enchondroma.
Fibrous Dysplasia
This idiopathic disorder is due to excessive proliferation of the spindle cell fibrous tissues in bones. Although 2 cases of a congenital autosomal recessive form of fibrous dysplasia have been reported, every other case has been sporadic, without any known hereditary component.
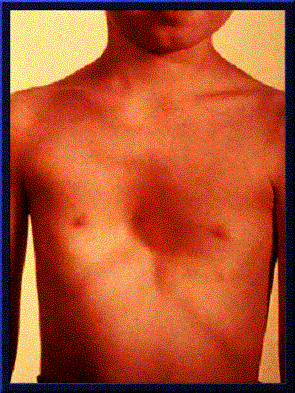
Clinical: The main clinical significance of this entity depends upon exactly which bones are affected. These bones will exhibit deformity, enlargement, and pain. The proximal femur is most commonly involved. The conventional form (Jaffe-Lichtenstein syndrome) may be monostotic or polyostotic. When accompanied by brown patchy skin lesions and endocrine abnormalities (especially precocious puberty) with multiple bone lesions, the condition is termed McCune-Albright syndrome.
Radiographic: Show a variable appearance with a highly lytic or ground-glass appearance; there is often a well-defined rim of sclerotic bone around the lesion. Although this process may occur rarely in the cortical bone, the vast majority of cases originate in the medullary space. Therefore, most cases present as bony enlargement with the process seeming to arise from an expanded medullary space.
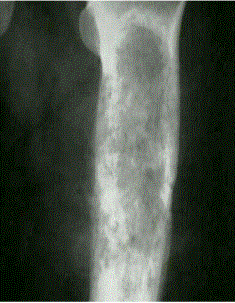
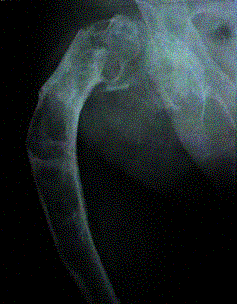
A. Characteristic ground glass appearance. B. Classic shepard’s crook deformity.
Pathology: The major histologic feature is proliferation of fibroblasts, which produce a dense, collagenous matrix. There are often trabeculae of osteoid and bone within the fibrous stroma. Cartilage maybe present in variable amounts. The bone fragments are present in a disorganized fashion and have been likened to “alphabet soup and “Chinese letters.”
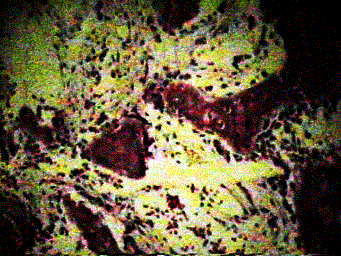
Prognosis: Overall good. Occasionally, pathological fractures will develop, and malignant transformation to osteosarcoma is seen rarely (< 0.5 %).
Treatment: Symptomatic. Internal fixation and bone grafting are used in areas of high stress where Nonoperative treatment would not be effective; most patients do not need surgical treatment.
Gaucher’s Syndrome
Inheritance: Aberrant AR lysosomal storage disease (also known as familial splenic anemia or sphingolipidosis). Commonly seen in children of Ashkenazic Jewish descent.
Frequency: 1:40,000
Clinical: Affected patients may complain of bone pain and occasionally experience a “bone crisis” (similar to sickle cell anemia). Bleeding abnormalities are also common. These patients may also be at increased risk for osteomyelitis.
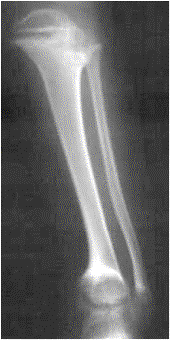
Radiographic: The marrow space in the bone is a closed space, so as these Gaucher cells enlarge, the intramedullary pressure begins to rise, which eventually may lead to occlusion of the intramedullary veins and hence bone infarction. As these bone infarcts evolve, one will be able to see the typical findings on MRI and then other imaging methods. The osteonecrosis may also develop in a subchondral location such as the femoral head in about half of patients, leading to subchondral collapse and early arthrosis. These patients may also exhibit other osseous findings, including the so-called “Erlenmeyer flask” deformities of the femoral metaphyses. These widened metaphyses may be seen in 40 – 50 % of patients, and may be due to the marrow packing of the Gaucher’s cells.
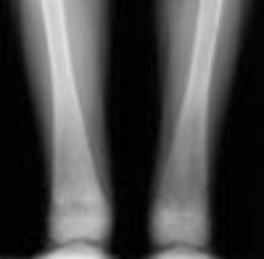
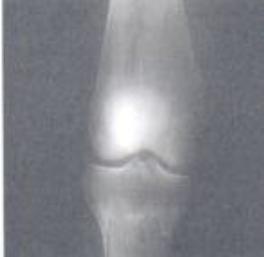
“Erlenmeyer flask” deformity of distal femur involving flaring of the metaphysis.
Pathology: Characterized by accumulation of cerebroside in cells of the reticuloendothelial system. Cause is a deficiency of the enzyme beta-glucocerebrosidase. The liver and spleen are usually quite enlarged. Histology demonstrates characteristic lipid-laden histiocytes (Gaucher’s cells) found in the bone marrow.
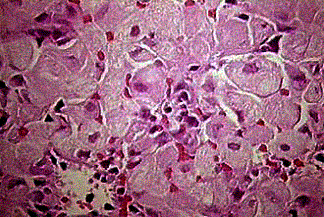
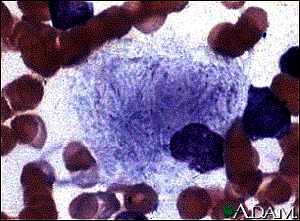
Prognosis: The usual form of the disorder is associated with a normal life span, although infantile and juvenile forms may result in mental retardation and an early demise.
Treatment: Basically supportive; new enzyme therapy is available, but extremely expensive.
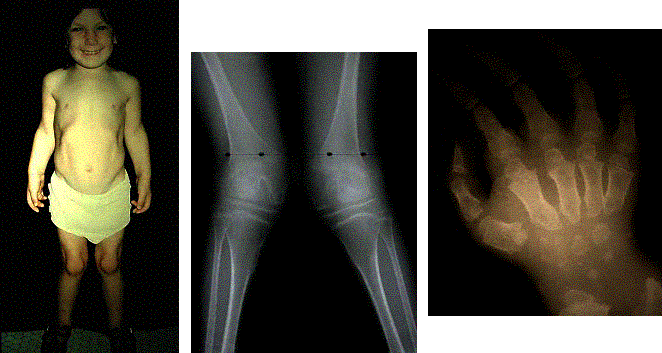
Hypophosphatemic Rickets
This has also been called X-linked hypophosphatemia, primary renal hypophosphatemic rickets or familial vitamin D-resistant rickets. As one of these names implies, it is due to a hereditary defect of the renal tubules, leading to decreased reabsorption of phosphate (phosphate diabetes) and therefore reduced serum phosphate levels. As the name also implies, this decreased reabsorption does not respond to usual amounts of vitamin D.
Inheritance: X-linked dominant. Mutations of the PHEX gene (phosphate-regulating gene with homologies to endopeptidases on the X chromosome) cause the disease. Most commonly encountered form of rickets.
Clinical: Laboratory studies reveal near-normal levels of calcium, PTH, and vitamin D, but low levels of serum phosphate. Urinary phosphate and serum alkaline phosphatase are elevated. Affected children present with bowing of the lower extremities, short stature, bone pain and dental caries. The disease becomes clinically apparent after 12 months of age.
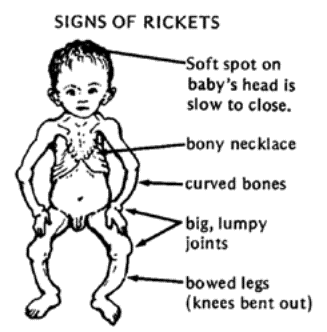
Classic beading of osteochondral junctions of ribs known as “rachitic rosary”
Radiographic: In general, this disorder exhibits rachitic epiphyseal and metaphyseal abnormalities predominantly in the lower limbs. This is best seen when comparing knee and wrist radiographs in the same patient. These patients also may demonstrate a generalized bone modeling error resulting in short, squat bones. In all cases of rickets, the study of choice is radiography of the wrists, knees, ankles, and long bones. No
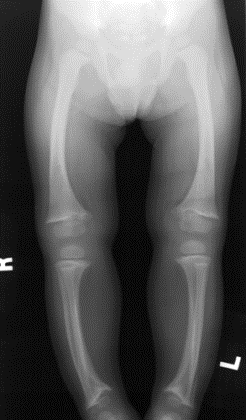
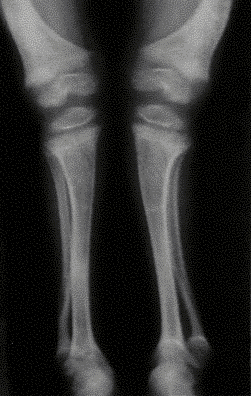
pathognomonic sign on x-ray distinguishes hypophosphatemic rickets from any other etiology.
Treatment: Treatment is primarily medical, typically consisting of oral replacement of phosphate and large amounts of vitamin D. Nephrocalcinosis is a known complication of medical treatment. Orthopedic management consists of osteotomies to treat residual deformities of the lower extremities. Careful preoperative planning is required because the deformities are multiplanar and thus may benefit from external fixation that precisely corrects angulation in each plane. Bone healing is delayed after osteotomy and may take approximately twice as long as expected in metabolically normal children.
Marfan’s Syndrome
A heritable disorder of fibrous connective tissue, Marfan syndrome shows striking pleiotropism and clinical variability. The cardinal features occur in 3 systems–skeletal, ocular, and cardiovascular.
Inheritance: AD. Although sporadic cases occur (about 25%), most cases are due to autosomal dominant gene with a high degree of penetrance. A paternal age effect is present, on average, in sporadic cases. All cases of the true Marfan syndrome appear to be due to mutation in the fibrillin-1 gene , which is located on chromosome 15.
Frequency: About 1:5000
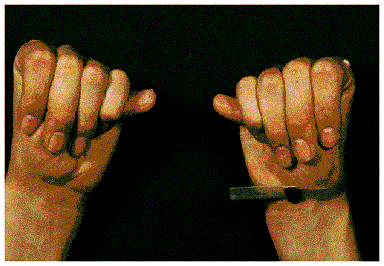
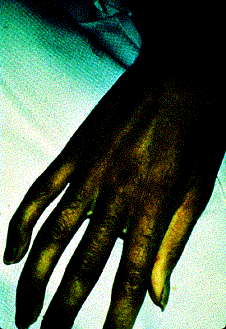
Clinical: These patients are characteristically tall and thin. The limbs are disproportionately long with respect to the trunk, especially in the hands and feet, giving the appearance of “arachnodactyly”. These subjective impressions of the patient may be objectified somewhat by looking for the “thumb sign” (the thumb protrudes beyond a clenched fist), and measuring the segmental index (distance from pubic symphysis to floor / distance from top of head to floor) and the metacarpal index (length / width).
Common ocular abnormalities include bilateral ectopia lentis, myopia, and retinal detachments. Associated cardiac abnormalities lead to a shortened life expectancy for these patients. These abnormalities include cystic medial necrosis of the aorta or pulmonary arteries (leading to dissection or rupture), aortic and mitral insufficiency, a “floppy valve” syndrome, and septal defects.
Radiographic: Other common skeletal findings include scoliosis and hypermobile joints. There is no typical scoliotic curve. Sagittal plane deformities are equally common and vary from hyperkyphosis to hypokyphosis.
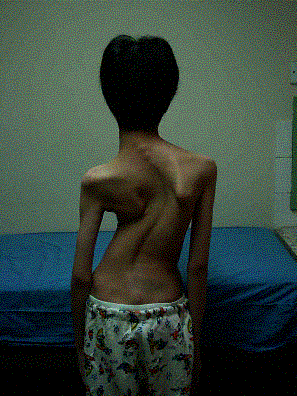
Prognosis: Improved cardiovascular management has greatly increased the life expectancy of patients with Marfan syndrome to nearly that of the general population.
Treatment: Use bracing for the same standard indications as in idiopathic scoliosis (although the success rate is lower)The presence of kyphosis with scoliosis requires anterior diskectomy and fusion with posterior fusion and instrumentation. Protrusio acetabuli can be treated with early triradiate cartilage epiphysiodesis.
Multiple Hereditary Exostoses
Multiple hereditary exostoses (EXT) is an autosomal dominant disorder characterized by multiple projections of bone capped by cartilage, most numerous in the metaphyses of long bones, but also occurring on the diaphyses of long bones. Flat bones, vertebrae, and the ribs may also be affected, but the skull is usually not involved. Deformity of the legs, forearms (resembling Madelung deformity), and hands is frequent.
Inheritance: AD. Penetrance is 100%, although severity of the disease varies. Multiple exostoses type I is caused by mutation in the gene encoding exostosin-1 (EXT1), which maps to chromosome 8q24. Multiple exostoses type II is caused by mutation in the gene encoding exostosin-2 (EXT2), on chromosome 11, and multiple exostoses type III has been mapped to a locus on chromosome 19 (EXT3). There is some evidence for an additional multiple exostoses locus
Frequency: 1:100,000
Clinical: Multiple bumps, which are usually nontender, may be palpable throughout the skeleton. The patient tends to be short in stature. The spine is rarely affected. Limb-length discrepancy and angular deformity are common because of disturbance of normal longitudinal growth. Scapular lesions can lead to “winging.”
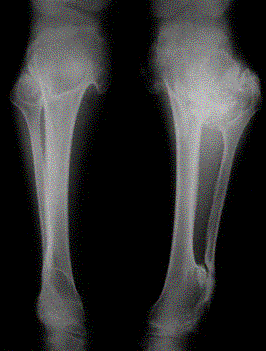
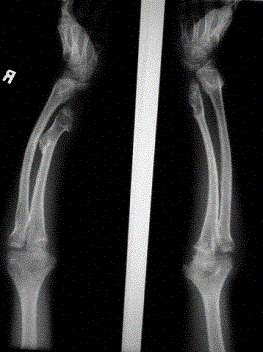
Radiographic: Osteochondromas usually have an absolutely pathognomonic appearance. The key word here is continuity. The cortex and medullary space of normal bone flows continuously into that of the osteochondroma (see figure below). Lesions can be seen around the metaphysis of the long bones, along the borders of the scapula, on the ribs, or originating from the iliac apophysis. The exostoses may be sessile or pedunculated. There is a predilection for the masses to appear at the end of the bone that grows the fastest and for greater involvement of the smaller bone of two-bone limbs (ulna & fibula). Pedunculated lesions typically point away from the epiphysis.
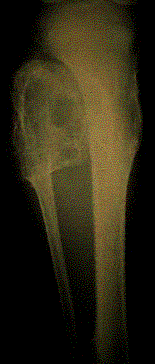
Pathology: Histologic examination reveals normal bone in the lesion that is in continuity with the metaphysis of the parent bone. The exostosis is covered by a cap of hyaline cartilage, usually less than 3 mm in thickness. The deep surface of the cartilaginous cap is involved in endochondral ossification, leading to growth of the lesion.
Prognosis: You can explain a lot of things about osteochondromas if you consider them to be an ectopic epiphysis. This means that they grow right along with the normal epiphyses, and stop growing when the plates close. Unfortunately, this syndrome is associated with an increased likelihood (probably up to 2-3 %, depending upon which study one quotes) that one or more of these osteochondromas will undergo malignant degeneration into something awful, usually a chondrosarcoma. There seems to be an increased propensity in those with a cartilaginous cap greater than 1 cm. Some patients without this syndrome will occasionally develop one or more osteochondromas. The likelihood of malignant degeneration is much lower (< 1 %) with sporadic osteochondromas such as this.
Treatment: Orthopaedic management consists of excision of painful exostoses and observation of those that remain asymptomatic. Angular deformities may require correction, especially at the knee, where valgus is common. Compression of surrounding structures may occur owing to the size of the lesions. The peroneal nerve is particularly prone to compression from masses and is susceptible to injury during surgical excision of proximal fibular lesions.
Once you have diagnosed this disorder, you must make sure that the patient knows the significance of their disorder and that they are now on a lifelong surveillance program. Any development of pain or growth after the plates have closed in an osteochondroma should be looked upon with suspicion for malignant degeneration. Follow-up imaging studies may include both radiographs and radionuclide images.
Neurofibromatosis
Neurofibromatosis is the most common single-gene disorder in humans.
Inheritance: AD, with 100% penetrance. There is a high rate of new mutations (almost 50%). Defect for NF1 found on chromosome 17, gene coding for neurofibrillin which acts as tumor suppressor.
Frequency: 1:3,000

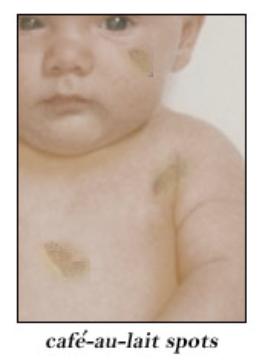
cutaneous neurofibromas
Clinical: There are several distinct forms of neurofibromatosis, two are common: the peripheral form (type I, von Recklinghausen syndrome — seen in 90 % of patients) and the central form (type II, familial acoustic neuroma).
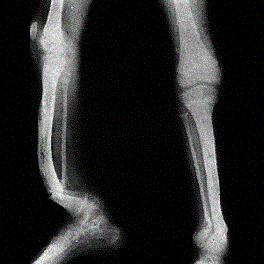
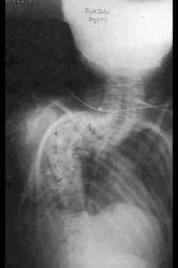
Virtually every part of the body is affected. Café au lait spots may be seen. The skeleton will be affected in about 80 % of patients. The most dramatic findings are the multiple neurofibromas seen throughout the body, especially in the peripheral, cranial, or spinal nerves and in the subcutaneous tissue. Fifty percent of patients with this disorder may develop kyphoscoliosis, usually in the high thoracic spine. Scoliosis is also common, although there is no standard curve pattern. This deformity may progress quite rapidly, and may lead to paraplegia. Other skeletal manifestations include posterior scalloping of vertebral bodies, hemihypertrophy (limb overgrowth), pseudarthrosis of the tibia (usually anterolateral), and enlargement of the spinal neural foramina.
Neurofibromatosis Type 1 Diagnostic Criteria (2 or more are diagnostic) 1. 6 or more café au lait spots (>5 mm prepubertal or >15 mm postpubertal)
2. 2 or more neurofibromas of any type, or 1 plexiform neurofibroma
3. Axillary and/or inguinal freckling
4. Optic nerve glioma
5. At least two Lisch nodules (hamartoma of the iris)
6. Osseous lesions, such as dysplasia of the sphenoid wing, vertebral scalloping thinning of long bone cortex, with or without pseudoarthrosis
7. First-degree relative (parent, sibling or offspring) with NF-1
Radiographic: There are a variety of radiographic anomalies of bone observed, ranging from a scalloping of the cortex to cystic lesions in long bones that look much like nonossifying fibromas, to permeative bone destruction. These radiographic findings can mimic benign and malignant bone lesions. Radiographs of the pelvis usually show various degrees of coxa valga, and in nearly 20% of patients there is radiographic evidence of protrusio acetabuli.
Pathology: Neurofibromas are composed of benign Schwann cells and fibrous connective tissue.
Prognosis: Survival is near normal. The exact incidence of malignancy in NF is controversial, with reported rates ranging from under 1% to over 20%. Types include neurofibrosarcoma, rhabdomyosarcoma of urogenital tract, Wilms’ tumor, childhood leukemia. 5-year survival for malignant sarcomas arising from fibromatoses is only 50%.
Treatment: Excision of neurofibromas is reserved for larger (>7 cm) lesions that are symptomatic or enlarging. The characteristic anterolateral bow is often obvious in infancy. Fracture usually follows with spontaneous union rare and surgical union a challenge. The bowed tibia should be managed with a total-contact orthoses to prevent fracture. IM rod fixation seems to offer the best results for the initial management of a pseudoarthrosis.
Osteogenesis Imperfecta
This inherited, generalized disorder of connective tissue is characterized by abnormal maturation of collagen. Recent basic science advances support the concept that OI is a series of syndromes representing classes of molecular defects, each with a reasonably well-defined clinical pattern. Most types of OI have been linked to mutations in type I collagen.
Inheritance: The majority of cases are inherited as AD or occur from new AD mutations. The mutations usually involve one of the two genes that encode the chains of type I collagen. COL1A1 found on chromosome 17 encodes the pro-a1(I) chain, and COL1A2 gene on chromosome 7 encodes the pro-a2(I) chain. Each type I collagen molecule contains two a1(I) chains and one a2(I) chain.
Frequency: 1:10,000
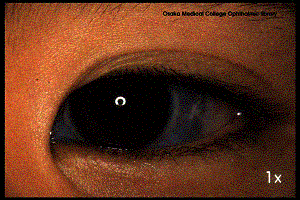
Clinical: It affects the skeleton, ligaments, skin, sclera, and teeth. The major clinical diagnostic triad is generalized osteoporosis with skeletal fragility, blue sclera, and odontogenesis imperfecta. Any two of these features suffice for the diagnosis.
Growth retardation occurs in most cases, and may be marked to the point of dwarfism in severe cases. This short stature is due to not only defects in collagen synthesis but also the cumulative fracture deformities secondary to the fragile bones.
This entity should be considered when one is entertaining the diagnosis of the battered child syndrome, another cause of multiple fractures in multiple stages of healing. The workup of a potentially battered child is extremely serious, and involves significant legal and social investigations of the parents. It would be tragic to mistakenly invoke this massively invasive process on a family by missing the findings of osteogenesis imperfecta. The take-home message: always look carefully for other classic signs of osteogenesis imperfecta, such as generalized osteoporosis, Wormian bones, blue sclera and odontogenesis imperfecta.
Sillence classification
Type I
Most common
AD
Blue sclerae
Mild; bone fragility (mostly preschool age fractures); hearing loss **
Type II
Very rare
AR
Blue sclerae (dark)
Lethal in perinatal period; crumpled long
bones; flattened vertebrae; beaded ribs
Type III
AR
White sclerae
Severe; short stature; fractures at birth and progressive deformity; spinal deformity
Type IV
AD
White sclerae
Moderate; bone fragility; no hearing loss; moderate growth failure **
**Type I/IV are divided into: A – without dentinogenesis imperfecta and B – with DI
Researchers have defined three more types of osteogenesis imperfecta (type V, type VI, and type VII), but the genetic causes have not yet been identified
Radiographic: The most common radiographic finding is that of generalized osteopenia. Multiple fractures resulting from insignificant trauma or normal muscle pull are also seen commonly, and may result in considerable deformity. Exuberant callus formation and pseudarthroses may also be seen. Persistent Wormian bones may be seen in the skull.
Pathology: Toward the more severe end of the spectrum of OI, bone often appears woven and only occasionally exhibits a lamellar pattern. The cortices are thin, with poorly developed haversian systems, and the trabeculae in the metaphyses are markedly attenuated. No change has been noted in the osteoclast population.
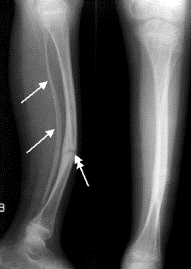
Prognosis: Depends on type. Type I may have little impact on patient. Type II is usually lethal. Types III and IV have a wide spectrum of involvement.
Treatment: As above, treatment depends on level of involvement. Several systemic treatment modalities have been attempted. Basic science and clinical literature support the use of pamidronate (bisphosphonate) in severe cases of OI, although it does not cure the disease. Management should begin with fracture prevention. Treatment for fractures in OI is often difficult. Fractures heal readily, often with exuberant callus, although the callus is also abnormal and easily deformed by early weight-bearing, leading to further deformities or shortening. Closed treatment is often used. Intramedullary fixation is superior to plates and screws, which tend to dislodge from the weakened bones. Management of deformity is often attempted with corrective osteotomies of larger bones. This is usually done with intramedullary fixation.
Scoliosis is often very difficult to treat. Curves tend to advance relentlessly and bracing is ineffective in controlling the progression of the deformity. Internal fixation is considerably hampered by the poor quality of the bone. Curves may be fused early (at 40 degrees) to halt the relentless progression.
Osteopetrosis
Also known as Albers-Schonberg disease, marble bone disease and osteosclerosis fragilis generalisata. This is another very logical disorder. The prime defect may be a failure of osteoclastosis. Without properly working osteoclasts, the whole bone remodeling process will fare badly, leaving one with short, weak, and oddly shaped bones. With abnormal osteoclasts, one might predict the following abnormalities:
Inheritance: 3 distinct forms described: AR (malignant and tarda forms) and AD (less severe). There exists evidence that a subset of autosomal recessive osteopetrosis is caused by mutation in the TCIRG1 subunit of the vacuolar proton pump. Autosomal recessive infantile malignant osteopetrosis can also result from mutations in the CLCN7 gene. Mutation has also been detected in the human homolog of the mouse ‘grey-lethal’ gene, also known as osteopetrosis-associated transmembrane protein-1 (OSTM1).
Frequency: Not well described, but roughly 1:200,000.

Clinical: Three clinically distinct forms of osteopetrosis are now recognized. Infantile-malignant for is fatal within the first several years of life without treatment. It manifests in infancy with thick, poorly remodeled dense bones. A proper medullary space is not created as the bones grow. Without a proper medullary space for the marrow, the patient will develop pancytopenia, leading to anemia (too few red cells), increased problems with infections (too few white cells), and bleeding problems (too few platelets). The child generally shows a failure to thrive, myelophthisic anemia and thrombocytopenia, hepatosplenomegaly, lymphadenopathy, spontaneous bruising and multiple fractures. Because of the abnormal remodeling, the neural foramina become small, causing encroachment and optic, oculomotor and facial palsies. There are no reports of any person surviving more than 20 years, usually from overwhelming infection or hemorrhage.
The intermediate form appears within the first decade of life and does not follow a malignant course. Some of the features noted in the malignant form, such as anemia, dental anomalies, or disproportion (short limb/short stature), can be identified in milder presentations.
The patient with the AD type has a normal life expectancy but many orthopaedic problems (this form is that also known as Albers-Schonberg disease). Mild anemia may be present; facial palsies and deafness can occur, but are not necessarily features. Fractures and subsequent deformities such as coxa vara are common.
Radiographic: The bones are overly dense in osteopetrosis patients. There may be transverse bands in the metaphyseal regions and longitudinal striations in the shafts. The metaphyseal regions, particularly the proximal humerus and distal femur, may develop a flask-shaped configuration. The pelvis may appear as a bone within a bone, and the sclerotic vertebrae may have a rugby jersey appearance.
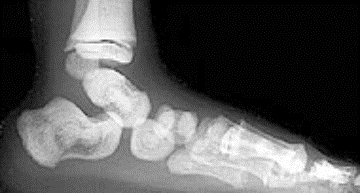
Pathology: Although generally considered a primary disorder of bone metabolism, with diminished bone resorption due to an osteoclast defect, studies indicate that osteopetrosis may more appropriately be considered an immune disorder resulting from a thymic defect that leads to the osteoclast abnormality. The histology of the bone shows that, in addition to thickened trabeculae and cortices, tongues of cartilage bars persist at the sites of endochondral bone formation, and may project far into the metaphysis and even into the diaphysis. The persistence of cartilage bars, normally resorbed by osteoclastic action in the zone of primary spongiosa, is characteristic of both rickets and osteopetrosis, but in osteopetrosis, the bars are calcified, and their central portions undergo osseous metaplasia. The bone is relatively hypocellular, with a paucity of osteoblasts and an almost complete absence of osteoclasts. The Subperiosteal new bone is in part nonlamellar, suggesting that intramembranous bone formation is also abnormal.
Treatment: Systemic treatment is an issue in the AR malignant form. High-dose 1,25-dihydroxyvitamin D coupled with a low calcium diet has been used because of its ability to stimulate osteoclasts and bone resorption. The malignant form has been treated successfully by allogeneic bone marrow transplantation from HLA-identical siblings, or by marrow ablation with cyclophosphamide and total body irradiation followed by marrow transplantation from a nonmatched donor. Treatment with recombinant interferon-g has been reported recently.
The orthopedic concerns are numerous. Fractures are common and require conventional treatment. Although healing does occur, time to healing can be prolonged. Coxa vara and long-bone deformity can result during the course of treatment of multiple fractures, both of which are amenable to corrective osteotomy. IM fixation is desirable, but can be difficult due to hardness of the bone and compromise of the marrow space. Osteomyelitis is common because of the diminished Vascularity and immune response.
Osteopoikilosis
This disorder is considered to be very common. It is characterized by small round or oval foci of bone sclerosis located in the trabecular bone, particularly in the pelvis, metaphyses and epiphyses of long bones, tarsals, and carpals. The shoulders, hips and sacrum are especially good places to look for these findings. These little deposits of bone are essentially multifocal bone islands. Although some disagreeable things have been associated with osteopoikilosis (subcutaneous nodules, osteosarcoma, spinal stenosis, osteosclerosis, etc. ) these associated findings are probably pretty darned rare.
Also known as Albers-Schonberg disease and osteopathia condensans disseminata.. The etiology and pathogenesis of osteopoikilosis is not clear. The sclerotomal distribution and association with abnormalities of mesodermal tissues suggests a relationship between this condition and other Osteosclerotic disorders.
Inheritance: AD. There is evidence that osteopoikilosis can an be caused by heterozygosity for loss-of-function mutations in LEMD3, also called MAN1, which encodes an inner nuclear membrane protein.
Frequency: Rare (not well described)
Clinical: The disorder develops during childhood and persists through life. Children have normal stature and most are asymptomatic, but up to 20% will have mild articular discomfort with a joint effusion. The diagnosis is often made as an incidental radiological finding. Fractures heal uneventfully and pathological fractures have not been reported. The main clinical significance is that these may be mistaken for sclerotic metastases.
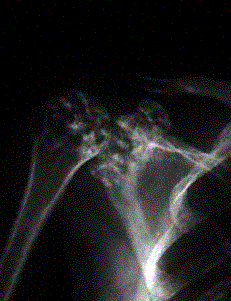
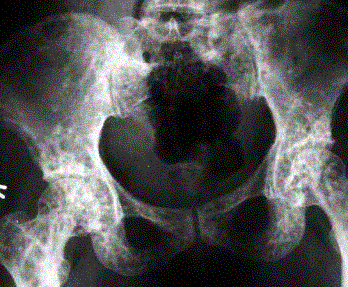
Radiographic: The osteosclerotic nodules are numerous well-defined, homogeneous, bilateral, circular- to ovoid-shaped from 1 to 15 mm, and are located in the metaphyses and epiphyses of long bones, the carpus, the tarsus, the pelvis, and the scapulae. They resemble cortical-like densities in cancellous bone. The ribs, clavicle and skull are not involved. Bone scan typically does not demonstrate increased uptake in the lesions, which is useful in differentiating this condition from metastatic breast or prostate carcinoma. Most of the time, their classic distribution and appearance will distinguish them readily from evil entities like mets.
Pathology: The sclerotic areas consist of focal condensations of compact lamellar bone within the spongiosa.
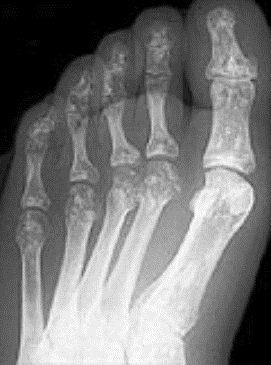
Prognosis: The risk of malignancy is probably not higher than the normal population. Osteopoikilosis is frequently seen in association with a hereditary dermatologic condition, dermatofibrosis lenticularis disseminata (Buschke-Ollendorf syndrome), which is marked by the presence of popular fibromas.
Treatment: There is no treatment for this benign disorder.
4. Could this dysplasia be in Taybi and Lachman’s Book?
Chances are extremely good that the syndrome is in Drs. Taybi and Lachman’s wonderful book, Radiology of Syndromes, Metabolic Disorders, and Skeletal Dysplasias. Besides providing a comprehensive list of syndromes, this book is also extremely well laid out. Each of the nearly 1000 entities in this book is listed the same way: its name, a list of synonyms, the mode of inheritance, frequency of occurrence, the clinical findings, the radiographic findings, a differential diagnosis (if any) and a list of pertinent references, including the original description(s) of the syndrome. This information is listed very efficiently (you have to, to get 1000 entities into only 800 pages). Thank goodness. When events have finally forced you to deal with the hand-foot-uterus syndrome once and for all, you pretty much just want to read the Cliff Notes version — not all 34 Cantos of Dante’s Inferno. Furthermore, this book is cross-referenced by means of lengthy gamut tables in the back of the book. If you can spot several key findings in the patient, you may be able to triangulate down onto a fairly short differential diagnosis or even one best diagnosis. If this process leads you to a certain diagnosis, life is good.
If you don’t have access to a copy of Taybi and Lachman’s book, you can go online to a wonderful website known as the Online Mendelian Inheritance in Man (OMIM) Database. This site is essentially a constantly-updated textbook on congenital disorders of all types. As of November 1, 2003, they listed 14,891 entries, which should provide a long enough differential diagnosis for almost anyone.
If you can’t find it in Taybi and Lachman or in OMIM, you have done your best, and it is probably time to punt.
Other that you’ll eventually hear about and see in your orthopaedic career are listed below. See nicely arranged table at the end of this section for a nice summary.
Spondyloepiphyseal Dysplasia
Spondyloepiphyseal dysplasia (SED) is a descriptive term for a group of disorders with primary involvement of the vertebrae and epiphyseal centers resulting in a short-trunk disproportionate dwarfism. Spondylo refers to spine, epiphyseal refers to the growing ends of bones, and dysplasia refers to abnormal growth.
Two major types of SED are recognized, namely, SED congenita and SED tarda. Spranger and Wiedemann first described SED congenita in 1966. Other rare forms of SED have been described. In 1973, Bailey suggested 2 groups in addition to SED congenita and SED tarda; these are pseudo-Morquio disease and pseudoachondroplasia SED.
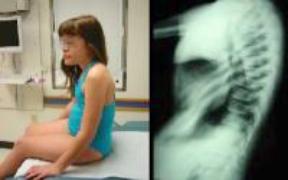
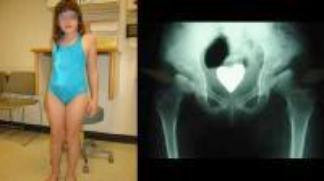
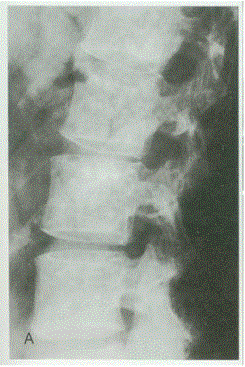
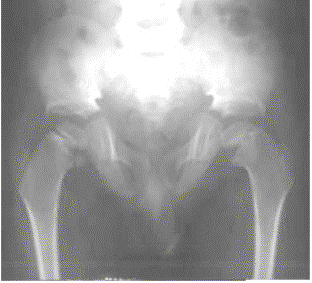
Clinical picture of a child with spondyloepiphyseal dysplasia. The child had a limp when she walked. The radiographs reveal Perthes-like changes. Both the hips appear to be in a similar stage of progression.
The sitting height is significantly affected. The trunk is disproportionately shorter than the extremities. The radiographs reveal platyspondyly
Inheritance: SED congenital is AD, SED tarda is usually XR (although it can be transmitted AD or AR. The gene for SED congenita has been mapped to the long arm of chromosome 12 (12q14.3), by mutations in COL2A1 (type II collagen alpha 1 chain) on chromosome 12. These result in abnormal type II collagen. Type II collagen is the major collagen of nucleus pulposus (spine), cartilage, and vitreous (eye).. Most cases result from random new mutations.
SED tarda is genetically distinct from SED congenital, caused by mutation in SEDL (SED late) gene. The SEDL gene has been identified on band Xp22. It encodes a protein of 140 amino acids with a role in vesicular transport.
Other skeletal dysplasias affected by collagen II include achondrogenesis type II, hypochondrogenesis, Kneist dysplasia, Stickler dysplasia, autosomal forms of SED tarda, and spondylometaepiphyseal (Strudwick) dysplasia.
Frequency: SED congenita is a rare genetic disorder. The prevalence is approximately 3.4 per million population. The incidence rate is approximately 1 per 100,000 live births.
Clinical: SED, metatropic dysplasia, and Kneist syndrome are considered short-trunk dwarfing conditions. SED is a generalized dysplasia with primary involvement of the vertebrae and proximal epiphyseal centers. SED congenita is a nonlethal form of congenital dwarfism characterized by typical skeletal dysplasias, vertebral changes, and ocular manifestations. It can be diagnosed at birth. In contrast, SED tarda is milder than SED congenita, late in onset, and appearance may be normal at birth.
SED congenita has variable severity. The face is generally taut, with a small mouth (frequently with a cleft palate). The trunk and extremities are shortened. There is frequently a pectus carinatum because the rib growth outpaces the trunk height. Scoliosis and kyphosis usually develop before the teen years. The hips are commonly in varus, the degree of which is the best marker for the severity of the disease. The knees are often in mild varus and the most common foot deformity is equinovarus. Medical problems include respiratory, ocular (often the most disabling) and occasionally hearing.
SED tarda features are often first brought to clinical attention about 4 years of age. Stature is mildly shortened. The condition may be first diagnosed as bilateral Perthes syndrome. Varus or valgus deformities are rare. Degenerative changes may occur in the hip or the knee by young adulthood.
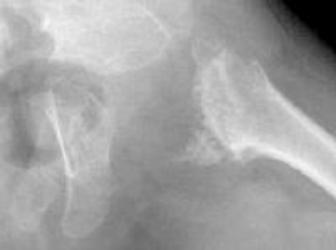
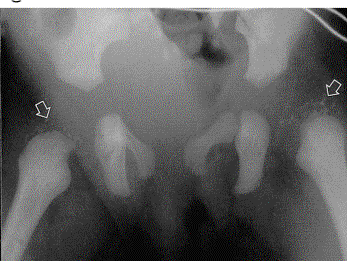
Delayed ossification of capital femoral epiphyses,
metaphyseal flaring, horizontal acetabular roofs, triangular fragment on the inferior aspect of the broad femoral neck, and coxa vara.
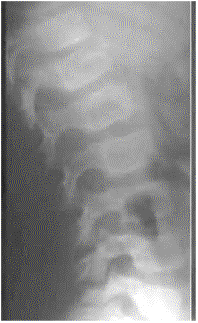
increased anteroposterior diameter, platyspondyly, posterior wedging of the vertebrae, and increased lumbar lordosis
Radiographic: Ossification is delayed in almost all regions. There is often odontoid hypoplasia or os odontoideum. Flattened vertebral ossification centers with posterior wedging give the vertebral appearance on the lateral view of a “pear shape.” The proximal femora are in varus with short necks, but varies. The proximal femur may not ossify for up to 9 years. Often the varus is progressive as is possible extrusion of the head. The distal femoral metaphyses are flared. Early osteoarthritis is likely in the hips, more so than the knee.
Pathology: Studies have indicated abnormal synthesis of type II collagen in SED congenita. Type II collagen is a primary matrix protein of physeal and epiphyseal cartilage. Abnormalities of the proliferative zone have been identified, with microcystic areas surrounded by a ring of cells. The chondrocytes of the resting zone appear vacuolated, containing periodic acid-Schiff (PAS)–positive cytoplasmic inclusions. Ultrastructural examination revealed these inclusions to be accumulations of fine granular material in dilated cisterns of rough endoplasmic reticulum. However, heterogeneity is present, and these findings are not consistent.
Prognosis: The standardized mortality ratio is not increased for patients with SED. Morbidity associated with SED may include the following conditions: neck instability (the most potentially serious problem); spinal deformities such as scoliosis, kyphosis, or lordosis; ocular abnormalities such as myopia or retinal detachment; hearing deficits; coxa vara, genu valgum, equinovarus foot
Treatment: A careful neuro exam should be done at each clinic visit (and flex/ext films every 3 years) to evaluate for possible cervical instability. It is recommended to fuse the AAI if instability exceeds 8 mm, or if symptoms develop. Surgical intervention of scoliosis is often necessary. Hip osteotomies are indicated if the neck-shaft angle is less than 100 degrees. Foot deformities can usually be treated according to standard clubfoot principles. If the foot is stiff, an osteotomy or decancellation of the talus, calcaneus, and/or cuboid may be needed.
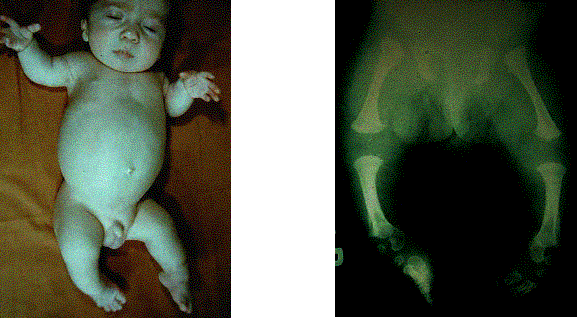
Chondrodysplasia Punctata
Also known as congenital stippled epiphysis and chondrodystrophia calcificans congenita. It has been subclassified into 3 groups: an XD-type (Chonradi-Hunermann syndrome), an AR rhizomelic type, which is usually fatal in infancy, and a rare XR type (four others have been described which are even more rare).
Inheritance: AR/XD, two forms. The AR type is a peroxisomal deficiency of dihydroxyacetone-phosphate acyltransferase. The genetic defect of the XD-type is thought to be caused by mutation in the gene encoding delta(8)-delta(7) sterol isomerase emopamil-binding protein.
Frequency: Approximately 1:100,000
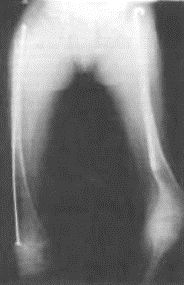
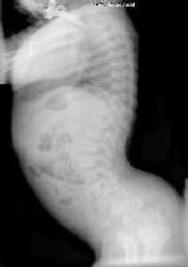
The above image is a autopsy radiograph and illustrates the lethal autosomal recessive form of the disease. The femora are shortened relative to the tibiae and fibulae.
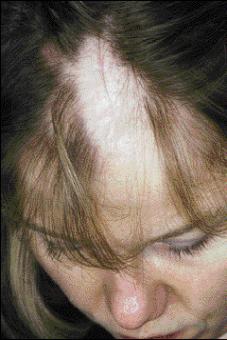
Hair is coarse and lusterless, often with areas of alopecia
Clinical: The severe AR rhizomelic form is usually fatal during the first year of life, often from respiratory causes or seizures. Findings include microcephaly, a high incidence of congenital cataracts and growth retardation. The XD form (Conradi-Hunermann) has a wide variation of clinical expression. Patients are characterized by hypertelorism, a depressed nasal bridge (not found in AR type) and a bifid nasal tip. Many often have alopecia, congenital heart and/or renal malformations and mental retardation. Asymmetric limb shortening and spinal deformities are common.
Radiographic: Characterized by multiple punctate calcifications seen on radiographs during infancy, although most disappear by 1 year. These involve the epiphyses, carpal bones, and pelvis. Although the appearance of neonatal epiphyseal calcification is striking, it is not very specific. Zellweger (cerebrohepatorenal) syndrome, gangliosidosis, rubella, trisomy 18 and 21, vitamin K deficiency, hypothyroidism, and fetal alcohol syndrome all may have the same phenomenon.
Other skeletal findings include limb-length inequality, coxa vara, and clubfoot or other foot deformities. Spinal findings include AAI, congenital scoliosis or kyphosis, often with hemivertebrae or congenital bars.
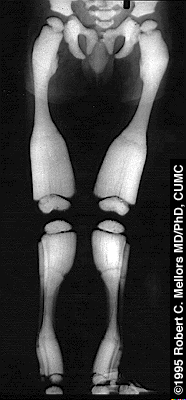
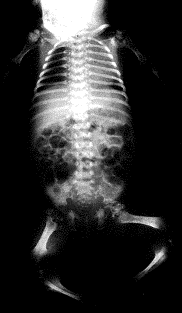
Multiple punctate calcifications are seen in the
epiphyses of the proximal femora.
Prognosis: As above, the AR form is usually lethal early. The AD form usually does not affect lifespan, but often has orthopaedic manifestations.
Treatment: Because of the risk of cervical instability, each patient should have a lateral cervical radiograph and, if instability appears possible, a flexion-extension view. The limb length discrepancies and spinal deformities often require surgical intervention. Coxa vara should be treated if the neck-shaft angle is less than 100 degrees.
Kneist’s Syndrome
Inheritance: AD, due to a defect in type II collagen. Most mutations are between exons 12 and 24 of the COL2A1 gene.
Frequency: 1:1,000,000 live births
Clinical: Short-trunked, disproportionate dwarfism with joint stiffness/contractures, scoliosis, kyphosis, odontoid hypoplasia and hypoplastic pelvis and spine. Joints appear enlarged because of broad metaphyses of the long bones, and they are stiff—often lacking both extension and full flexion. Respiratory problems and cleft palate are common. Associated retinal detachment and myopia require an ophthalmology consult. Otitis media and hearing loss are common. Intellectual development is normal.
Radiographic: Radiographs show osteopenia of the spine and extremities. All regions of the spine are affected, from AAI to hypoplasia of the cervical vertebrae and flattening of all vertebrae. Vertebral bodies have vertical clefts. The femoral necks are short and broad. Valgus deformities often develop in the distal femur or proximal tibia.
Pathology: The cartilage has been termed “soft and crumbly with a “Swiss-cheese” appearance. May be related to an abnormality of cartilage proteoglycan metabolism, and physes also may have the characteristic “Swiss cheese” appearance histologically.
Treatment: Early therapy for joint contractures is required. Reconstructive procedures may be required for early hip degenerative arthritis. It is important to rule out cervical instability when the diagnosis is first made, when intubation is planned or with any loss of milestones or of strength or coordination. Surgery for spinal deformity is not often needed.
Metaphyseal Chondrodysplasia
Heterogeneous group of disorders characterized by metaphyseal changes of tubular bones with normal epiphyses. The name refers to the end result (radiographic changes in the metaphyses), but the defect is in the growth plate itself. There are many different named disorders that come under the heading of metaphyseal chondrodysplasia. Listed below are the most common types.
Inheritance: McKusick (also known as cartilage hair hypoplasia–CCH): AR; Schmid, Jansen, and Kozlowski: AD
Frequency: In the Amish, the incidence of CHH is 1.5 in 1,000births. CHH is also found in Finland at a high frequency, approximately1 in 23,000 births
Clinical: Several types are recognized, including the following:
1. Jansen’s (rare) – AD, due to defect in parathyroid hormone receptor and parathyroid hormone related protein. This is the most severe form–mentally retarded, markedly short-limbed dwarf with wide eyes, monkey-like stance, and hypercalcemia. Striking bulbous metaphyseal expansion of long bones is a distinctive radiographic finding.
2. Schmid’s – AD, defect is in the a1 chain of type X collagen (restricted only to hypertrophic chondrocytes in the calcifying zones of the growth plate and in zones of secondary ossification). More common, less severe form. Short-limbed dwarf not diagnosed until patient is older, with stunting of growth and bowing of legs due to coxa vara and genu varum. Predominantly involves the proximal femur. Metaphyseal lesions heal with bed rest but recur with weight bearing. Often confused with rickets, but lab tests are normal.
3. McKusick’s – AR, mapping to chromosome 9, although etiology has not been further determined at this point. Hypoplasia of cartilage and fine, thin, brittle hair are notable features. Most common among the Amish population and in Finland. Atlantoaxial instability is common (odontoid hypoplasia) and requires flexion-extension lateral radiographs for proper evaluation. Ankle deformity develops due to fibular overgrowth distally. May have abnormal immunologic incompetence (susceptible to severe reaction from chickenpox)
Radiographic: McKusick: there is more shortening and less varus of the long bones than in the Schmid type. The metaphyseal involvement is more evenly distributed, not just medial. AAI has been reported. The TL spine shows some minimal changes, which are not of much clinical importance. Schmid: The metaphyses of the long bones are widened and flared, and may have cysts. The physes are slightly widened. There is varus deformity of the knees. AAI has been reported, but is rare. Jansen: More severe metaphyseal changes.
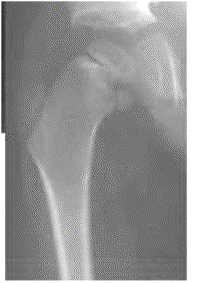
Pathology: The physis (proliferative and hypertrophic zones) histologically appears to be more affected than the metaphysis; therefore, the term metaphyseal dysostosis has fallen out of favor. The epiphyses are normal.
Prognosis: Those with milder types of immune deficiency have lived to adulthood, some even to old age. Note, however, the severity of the immunodeficiency varies; in one series, 11 of 77 patients died before age 20 yr but two were still alive at age 76.
Treatment: Rule out rare AAI correct genu varum if severe and monitor medical problems in McKusick type
Multiple Epiphyseal Dysplasia
MED is one of the most widely known and commonly occurring skeletal dysplasias. If affects many epiphyses, produces symptoms mainly in those with significant loadbearing, and has few changes in the physes or metaphyses.
Inheritance: Most forms AD, although at least one form described as AR. Some have defect in cartilage oligomeric matrix protein (COMP), others in type IX collagen
Frequency: Not well described.
Clinical: Short-limbed disproportionate dwarfism that often does not manifest until age 5-14. Must differentiate from Spondyloepiphyseal dysplasia. A mild form (Ribbing’s) and a more severe form (Fairbanks’) exists. They may be referred for joint pain, decreased range of motion, gait disturbance, or angular deformities of the knees. There may be flexion contractures of knees or elbows. The spine and face are normal except for occasion hypoplasia of the odontoid can produce cervical instability and subsequent neurologic loss. There is no visceral involvement.
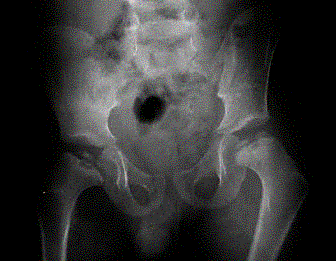
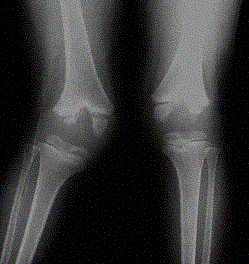
Radiographic: Most changes in MED involve the epiphyses; almost all of the ossification centers are delayed in appearance. There are occasional irregularities of streaking in the metaphyses. In the growing patient, the epiphyses are fragmented and small in size. The ossification centers eventually coalesce, but the overall shape is smaller. The more fragmentation there is in the capital femoral epiphysis, the earlier onset of osteoarthritis. Coxa vara occurs in some patients. In adulthood, major joints develop premature osteoarthritis, which is most common and severe in the hips. Short, stunted metacarpals/metatarsals, abnormal ossification (tibial “slant sign” and flattened femoral condyles), valgus knees, and waddling gait are common. The proximal femoral involvement can be confused with Perthes’; MED is bilateral and symmetric, has early acetabular changes, and does not have metaphyseal cysts.
Pathology: There is a failure of formation of the secondary centers of ossification causing a loss of articular cartilage support with resulting joint deformity and early arthritis. Growth plate organization is still noticeably abnormal, despite the minimal changes seen in the metaphyses. Mutations have been found in the gene for COMP on chromosome 19 (as in pseudoachondroplasia), but also on COL9A2 (coding for a22 fibers of collagen IX)
Prognosis: Not all patients need surgical treatment due to a wide spectrum of disease, but due to the propensity for early degenerative disease, many patients will go on to require joint replacement.
Treatment: Observations versus acetabular coverage in childhood, as well as occasional realignment procedures. Joint replacement in adulthood. C1-C2 fusion for instability.
Mucopolysaccharidosis
The group of clinical conditions produced by a common inability to metabolize one of several proteoglycans (mucopolysaccharides) is called mucopolysaccharidosis. The various mucopolysaccharides accumulate with different tissue affinity: some affect the CNS, some the eyes, and some the skeleton. All are inherited as recessive states and all but Hunter’s syndrome (sex-linked) are autosomal. Most are differentiated by clinical exam, x-ray, and qualification and quantification of the various MPS in the urine.
Hurler’s syndrome – gargoylism – AR, deficiency in alpha-L-iduronidase
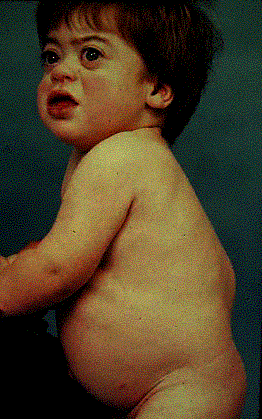
Clinical presentation:
-usually noted shortly after birth
-gargoyle appearance with thickened lips, wide nostrils, and large ears
-mental retardation
-elbow, hip and knee flexion contractures
-enlarged liver and spleen
-impaired vertical growth
-corneal clouding
-Lab: increased urine heparan and dermatan sulfate
Radiographs:
-slipper shaped sella turcica
-spine most characteristic with anterior-inferior vertebral beaking
-short leg bones
-wide metacarpals
Treatment:
-patients rarely survive beyond adolescence
-some patients have benefited from bone marrow transplant
Hunter’s syndrome – similar to Hurler’s except less severe and slower to progress
Clinical presentation:
-differentiated by no corneal clouding and only males affected (XR)
-Lab: increased urine heparan and dermatan sulfate
San Filippo’s syndrome – AR, often misdiagnosed as CP
Clinical presentation:
-all features begin in early childhood and are progressive
-behavioral problems
-hyperreflexia and spasticity
-progressive mental retardation
-face has thick, bushy eyebrows
-usually bedridden and unaware of time or place by adolescence
-Lab: increased urine heparan sulfate
Morquio’s syndrome – autosomal recessive
Clinical presentation:
-usually noted between 18 to 24 months, present with waddling gait, knock
knees, thoracic kyphosis, and pectus carinatum
-growth stunted, mainly in the trunk
-progressive genu valgus
-normal intelligence (only MPS without some mental retardation)
-Lab: increased urine keratan sulfate
Radiographs:
-spine: platyspondyly with tongue deformity of vertebral body seen on
lateral film, kyphosis is common
-pelvis: coax vara with oversized acetabulae
-hands: tapering “pencil shape” of proximal metacarpals
-c-spine: hypoplastic odontoid with C1-C2 instability
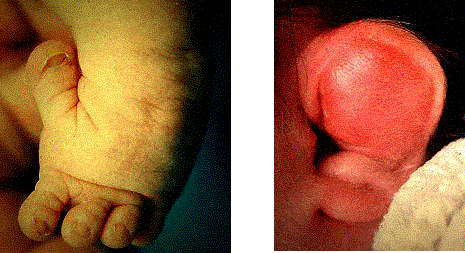
Diastrophic Dysplasia
Inheritance: AR, failure of DTD gene on chromosome 5 which encodes a sulfate transporter protein, leading to under-sulfation of cartilage proteoglycans
Frequency: The disorder is extremely rare, except in Finland, where between 1 & 2% of the population are carriers.
Clinical: Severe, short-limbed dwarfism. This “twisted” dwarf classically has a cleft palate, severe joint contractures (especially hip and knees), cauliflower ears, hitchhiker thumb (and great toe), rigid club feet, mid-thoracic kyphosis, cervical kyphosis, thoracolumbar kyphoscoliosis, spina bifida occulta and atlantoaxial instability due to odontoid hypoplasia. The joints can be dislocated, especially the shoulder, elbows, hips, and patellae (knee caps). Flexion contractures of knees and shoulders are common.
Scoliosis is not present at birth but often is progressive, especially in the early teens.
Radiographic: Irregular epiphyseal ossification, dislocated hips and cervical kyphosis. In infancy, calcification develops in the pinna of the ear, and later in the cranium and costal cartilages. The vertebrae are poorly ossified. The lower cervical spine may demonstrate kyphosis. Only one case of AAI has been reported. The first metacarpal is small, oval and proximally placed. Both the ulna and fibula are shortened, contributing to the valgus of the knees and the radial head subluxation, which are sometimes seen. The diaphyses of the long bones are short and broad.
Pathology: Associated with a disorder of type II collagen in the physis with failure of formation of the secondary centers of ossification and severe soft tissue contractures. Chondrocytes appear to degenerate prematurely, and collagen is present in excess.
Prognosis: There is increased mortality in infancy due to respiratory complications but thereafter, people with diastrophic dysplasia have a normal life span.
Treatment: The cervical kyphosis is often severe and requires immediate treatment. Surgical release of club foot deformities, osteotomies for contractures, and spinal fusion are often required. No treatment of hip dislocations.