
Osteoarthritis (OA), also known as degenerative joint disease (DJD) is the final common pathway to which all joint disease deteriorates. Post traumatic, septic and other inflammatory arthropathies have components of DJD in their end stages. OA imposes an enormous disease burden currently affecting approximately 40 million people in the U.S. and is expected to impair 60 million Americans by the year 2020. This disease is a leading cause of disability and impaired quality of life. The classic symptoms of OA are characterized as pain, stiffness, and loss of mobility due to degeneration of synovial joints. As DJD progresses, crippling deformity and joint instability ensue. (1)
Degenerative joint disease can be divided into two subgroups, primary and secondary DJD. Secondary osteoarthritis has a well defined etiology contributing to joint destruction such as trauma or inflammation while primary OA is multi-factorial. Degenerative joint disease (DJD), regardless of etiology, implies the progressive loss of articular cartilage in the setting of inadequate cartilage repair. This results in progressive pain, stiffness and deformity as well as a remodeling of subchondral bone. Traumatic and degenerative lesions to articular cartilage do not remodel into native type II collagen enriched cartilage, but a rather inferior tissue known as fibrocartilage. This is a fundamental and contrasting difference from the remodeling seen in osseous lesions. Fractures and other lesions in bone have the potential to ultimately form healthy bone while cartilage will remodel into inferior tissue if it remodels at all. Inflammatory arthropathies result in joint destruction through the action of lytic metalloproteinases and collagenases released during the inflammatory cascade. Inflammatory arthropathy has many inciting etiologies including autoimmune, crystalline and septic sources, all of which can initiate catastrophic inflammation. Whatever the mechanism, once cartilage has been destroyed it has little capacity to regenerate a healthy robust articular surface. This biologic phenomenon is an underlying reason why arthroplasty has revolutionized the practice of orthopaedic surgery over the past four decades with artificial joint resurfacing.
The main functions of articular hyaline cartilage are: 1)To provide a shock-absorbing structure which can withstand compression, tension and shearing forces, 2) Dissipate highly repetitive loading forces {Hyaline cartilage in the knee has to deal with repetitive mechanical forces that can sometimes reach 65 times body weight.}, and 3) To provide an almost frictionless articulating surface. Interestingly, no known substance can do this as well. In the absence of any seditious event, cartilage can function well for a lifetime of use, although age-related changes do occur.
Because articular cartilage is aneural and avascular, articular chondrocytes depend solely upon soluble factors in its matrix (i.e. protein growth factors) and mechanotransduction (molecular signaling from mechanical forces applied to cells) to interpret their environment. Cartilage nutrition is therefore primarily from the synovial fluid and to some extent from the adjacent bone. This has implications with regard to healing since superficial lesions rely solely on synovial nutrition and don’t have the direct support of underlying endosteal bone. Deep articular lesions, which reach the underlying bone and blood supply, heal better because of their direct access to repair cells and nutrients found in the endosteal blood supply. This is the rational for arthroscopic micro-fracture techniques that cause cracks in the underlying subchondral bone and allow stem cells and nutrients to jump start the repair of a smoldering superficial lesion. This type of cartilaginous repair, although better than a gaping defect, is far from ideal since the type of cartilage produced is not the original hyaline cartilage but ‘fibrocartilage’. Fibrocartilage is biomechanically inferior to the real deal and can lead to precocious joint deterioration.
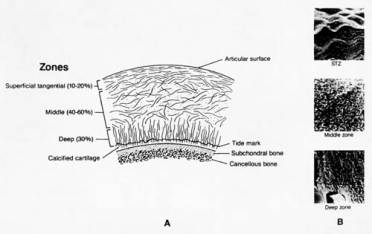
Cartilage is made up of four zones that function together to perform the functions mentioned above. These zones include 1)superficial tangential zone (STZ), 2)middle zone, 3) deep zone, and 4) calcified zone. (Figure 1) The cells of the superficial zone have an ellipsoidal shape and lie with their long axes parallel to the articular surface. The cells of the other zones have a more spheroidal shape. In the deep zone, they tend to align themselves in columns perpendicular to the joint surface.
Figure 1
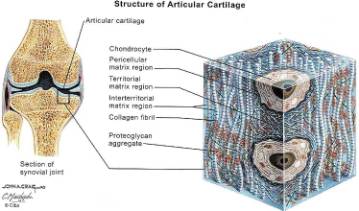
Hyaline cartilage typically is composed of by weight: 70% water, 15% collagen (Type II collagen makes up 90% to 95% of the total collagen fraction), 15% proteoglycans (GAGs). Type II collagen imparts cartilage with strength to resist tensile stress while GAGs with their hydrophilic side groups mainly resist compression. These components are distributed among the chondrocytes, non-collagen proteins, lipids and inorganic material found in articular cartilage. (Figure 2,3) Similar to bone, cartilage constantly undergoes dynamic remodeling. Cartilage is eroded by matrix metalloproteinases (MMP’s) while it is being reformed by chondrocytes and chonroblasts.
Figure 2
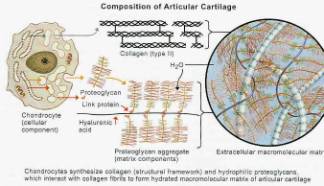
Figure 3
Despite the vast prevalence of primary osteoarthritis, until recently, relatively little has been known about the actual etiology of primary OA. Also confusing is the fact that the clinical symptoms of OA often do not correlate with their corresponding radiographic lesions. Epidemiological studies show wide variations in physical activity, diet and medical history in leading to patients who present with a widespread range of symptoms and degenerative lesions. Fewer than one half of patients with radiographic evidence of osteoarthritis have significant clinical symptoms. Therefore, treatment is primarily predicated upon symptoms rather than on the degree of visible pathology. Nevertheless, the root problem remains the imbalance of articular cartilage destruction over its synthesis. (1)
Current theories in OA begin to describe the molecular details behind the imbalance of cartilage degradation and reformation. The initial response of a diseased joint is to induce proliferation in chondrocytes, and increase collagen and proteoglycan synthesis. However this takes place in the setting of increased metalloproteinase (MMP) activity (the enzymes involved in denaturing cartilage). As the disease progresses, the ability of chondrocytes to repair the articular surface is outstripped by progressive cartilage degradation. Fibrillation, erosion and cracking initially appear in the superficial layer of cartilage and progress over time to deeper layers, resulting in large clinically observable erosions. (Figures 4, 5, 6, 7, 8) Chondrocyte loss, via apoptosis, is induced by nitric oxide (NO), which itself is produced by pro-inflammatory stimuli such as interleukin 1 (IL-1) and tumor necrosis factor a (TNF-a). Interleukin-1 (IL-1) is a pro-inflammatory molecule key in the development of both OA and rheumatoid arthritis. IL-1 has the following actions: 1)Stimulates the synovial cells to produce more metalloproteinases, 2)Inhibits the synthesis of type II collagen in articular cartilage, 3)Inhibits the synthesis of proteoglycans by chondrocytes. 4) Induces chondrocytes to produce disorganized cartilage matrix proteins. Therefore IL-1 not only causes degradation of cartilage, but suppresses any anabolic attempt to repair it. (For a busy but illustrative summary see Figure 12)
Figure 4
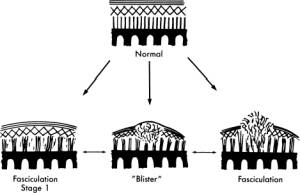
Figure 5
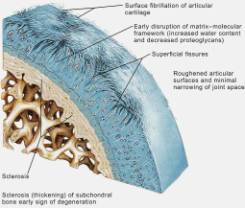
Figure 6
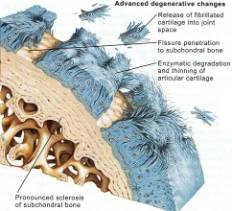
Figure 7
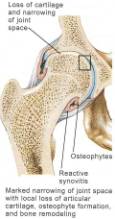
Figure 8
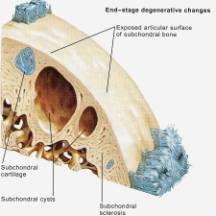
Figure 9
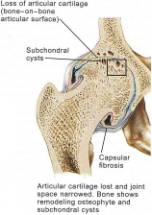
For many years OA was simply thought to be an inevitable consequence of aging, the “wear and tear” phenomena. However some elderly patients, despite a lifetime of being very active never develop OA. A new understanding is emerging about the natural history of primary DJD. The articular cartilage matrix generally undergoes important structural, mechanical, and molecular changes with age. These include surface fibrillation, alteration of proteoglycan structure and composition, increased collagen cross-linking, and decreased tensile strength and stiffness.(8,9) Deterioration of chondrocyte function accompanies these matrix changes. With age, chondrocytes synthesize smaller aggrecan molecules and less functional link proteins, leading to the formation of smaller, more irregular proteoglycan aggregates.(10) The mitotic and synthetic activity of human chondrocytes declines with age. Human chondrocytes become less responsive to anabolic mechanical and humoral signals. Given these age-related biological changes in chondrocytes, it is not surprising that there is an increase in the incidence of presenting OA with each passing decade. After the age of fifty, the risk of posttraumatic osteoarthritis following an intra-articular fracture of the knee increases 3 to 4 fold. (11) Interestingly, techniques like microfracture, perichondrial arthroplasties, and drilling of articular surfaces are much less effective in patients more than fifty years of age compared to patients less than thirty years of age. (12) Many of these observations are thought to be due to age-related chondrocyte senescence, inducing an age-related deterioration of cell function and replicative capacity. (13)
Some evidence exists that OA has a genetic predisposition (3). The presence of Heberden’s nodes signifies a predisposition toward the development of osteoarthritis, and this may potentiate the effects of local etiologic factors, such as trauma, inflammation, or instability. Interestingly, patients with meniscal tears who also have Heberden’s nodes are more likely to develop posttraumatic arthritis of the knee after meniscectomy (4). Other risk factors of osteoarthritis are obesity, increased bone density, trauma, and repetitive stress (5).
Multiple studies have shown the importance of motion, in excess or deficiency, on the health of articular cartilage. (6) The Framingham study showed the risk of developing OA was greater in patients who had a profession requiring heavy labor with or without obesity. Nevertheless, several studies have shown that development of obesity correlates of with development OA. Interestingly, biomechanical studies on canine beagle articular cartilage showed that running dogs 4 km a day resulted in thicker more biomechanically viable articular surface compared to rested dogs, while running the dogs more than 20 km a day resulted in increased articular wear and decreased cartilage biomechanical stiffness compared to rested dogs. This suggests a possible dose response effect to activity. Additionally, multiple studies have demonstrated how activity or cyclic loading of cartilage results in a marked increase in expression of chondrocyte glycosaminoglycans (GAGs) and type II collagen. Glycosaminoglycans (GAGs) and type II collagen are the molecules responsible for compressive and shear strength of articular cartilage respectively. In normal articular cartilage these structures are latticed together in the territorial matrix of chondrocytes (fig 2, 3) and interact directly with cellular adhesion molecules on the chondrocytes surface. Interestingly, if the same cartilage explants that were previously induced by cyclic loading are subjected to static loading (a maneuver akin to immobilizing a joint in a cast), the synthesis of GAGs and type II collagen plummets in comparison to control tissue that was neither statically or cyclically loaded. Whatever the actual molecular mechanisms, it appears that mechanotransduction has a significant role in articular cartilage health as well as the basic modulation of chondrocytes.
In the early stages of osteoarthritis, there is actually an increase in thickness owing to increased water content (swelling) and an increase in the net rate of synthesis of PG. The normal water trapping structures of glycosaminoglycans (Figs 2, 3) become disorganized and shortened with DJD and therefore become less efficient at compressive shock absorbing despite their increased synthesis. This attempt to repair articular surfaces may last for years in humans (7). Cartilage repair is dependant on the production of matrix by chondrocytes which is stimulated by anabolic growth factors like Insulin like growth factor-1 (IGF-1), Transforming Growth Factor B (TGF-B) and Basic Fibroblastic Growth Factor (B-FGF) as well as by mechanotransduction. IGF-1 and cyclic mechanical loading have been shown to have a synergistic anabolic response on articular chondrocyte function in-vitro. (14) With OA disease progression, the joint surface thins and the PG content decreases as enzymatic and traumatic degredation outstrip the articular capacity for increased synthesis. Progressive fibrillation of the cartilage occurs, and eventually, the underlying bone is exposed. (Figs 4,5,6) As the articular surface is worn away, an osteoblastic response concurrently ensues. Articular chondrocytes, like those in the growth plate, represent different pathways of terminal differentiation that can be redirected towards endochondral bone formation. In DJD articular chondrocytes recapitulate events of endochondral bone formation driving subchondral sclerosis. Penetrating synovial fluid and mechanical signals trigger an osteoblastic response, like that seen after a fracture, which is manifested by marginal sclerosis and osteophytes. (Fig 10, 11) Proteins in the synovium activate receptors on periarticular chondrocytes and osteoblasts resulting in the gene expression that drives the proliferative response. Erosive pressure from invading synovial fluid also causes the formation of peri-articular bone cysts. (Fig 9) Osteophytes together with the thickening of the joint capsule, lead to limitation of motion and deformity. (Fig 10, 11)
Figure 10
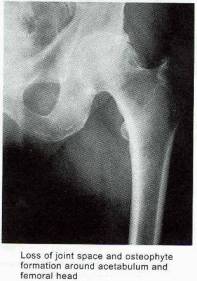
Figure 11
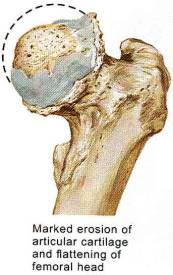
Note the dotted line is the correct anatomic position of the non-degenerative femoral head, as well as the significant osteophytes produced during this productive and erosive process.
Figure 12

This busy figure summarizes many of the inflammatory and enzymatic signals involved in DJD and particulate disease.